







Rapport de l'OPECST n° 185 (2004-2005) de M. Claude SAUNIER , fait au nom de l'Office parlementaire d'évaluation des choix scient. tech., déposé le 15 février 2005
Disponible au format Acrobat (1,3 Moctet)
-
PRÉFACE
-
INTRODUCTION GÉNÉRALE
-
PREMIÈRE PARTIE :
LA SÉCURITÉ SANITAIRE : ÉTAT DES LIEUX
-
DEUXIÈME PARTIE :
L'AFSSA ET LA SÉCURITÉ SANITAIRE DES ALIMENTS
-
TROISIÈME PARTIE :
L'AFSSAPS ET LA SÉCURITÉ DES PRODUITS DE SANTÉ
-
QUATRIÈME PARTIE :
UNE CLARIFICATION NÉCESSAIRE
-
RECOMMANDATIONS
-
EXAMEN DU RAPPORT PAR L'OFFICE
-
LISTE DES PERSONNES AUDITIONNÉES
|
N° 2108 ___ ASSEMBLÉE NATIONALE CONSTITUTION DU 4 OCTOBRE 1958 DOUZIÈME LÉGISLATURE __________________________________ Enregistré à la Présidence de l'Assemblée nationale Le 18 février 2005 |
N° 185 ___ SÉNAT SESSION ORDINAIRE DE 2004 - 2005 ________________________________ Annexe au procès-verbal de la séance du 15 février 2005 |
|
OFFICE PARLEMENTAIRE D'ÉVALUATION
DES CHOIX SCIENTIFIQUES ET TECHNOLOGIQUES
________________________
RAPPORT
sur
L'APPLICATION DE LA LOI N° 98-535 DU 1 ER JUILLET 1998
RELATIVE AU RENFORCEMENT DE LA VEILLE SANITAIRE
ET DU CONTRÔLE DE LA SÉCURITÉ SANITAIRE DES PRODUITS DESTINÉS À L'HOMME
Par M. Claude SAUNIER,
Sénateur
|
_________
Déposé sur le Bureau
par M. Claude BIRRAUX, Premier Vice-Président de l'Office |
_________ Déposé sur le Bureau du Sénat par M. Henri REVOL, Président de l'Office |
|
_______________________________________________________________________
Composition de l'Office parlementaire d'évaluation
des choix scientifiques et technologiques
Président
M. Henri REVOL
Premier Vice-Président
M. Claude BIRRAUX
Vice-Présidents
M. Claude GATIGNOL, député M. Jean-Claude ÉTIENNE, sénateur
M. Pierre LASBORDES, député M. Pierre LAFFITTE, sénateur
M. Jean-Yves LE DÉAUT, député M. Claude SAUNIER, sénateur
|
D ÉPUTÉS |
SÉNATEURS |
|
M. Jean BARDET M. Christian BATAILLE M. Claude BIRRAUX M. Jean-Pierre BRARD M. Christian CABAL M. Alain CLAEYS M. Pierre COHEN M. Francis DELATTRE M. Jean-Marie DEMANGE M. Jean DIONIS DU SÉJOUR M. Jean-Pierre DOOR M. Pierre-Louis FAGNIEZ M. Claude GATIGNOL M. Louis GUÉDON M. Christian KERT M. Pierre LASBORDES M. Jean-Yves LE DÉAUT M. Pierre-André PÉRISSOL |
M. Philippe ARNAUD M. Paul BLANC Mme Marie-Christine BLANDIN Mme Brigitte BOUT M. François-Noël BUFFET M. Roland COURTEAU M. Jean-Claude ÉTIENNE M. Christian GAUDIN M. Pierre LAFFITTE M. Serge LAGAUCHE M. Jean-François LE GRAND Mme Catherine PROCACCIA M. Daniel RAOUL M. Ivan RENAR M. Henri REVOL M. Claude SAUNIER M. Bruno SIDO M. Alain VASSELLE |
SIGLES ET ABRÉVIATIONS
AESA Agence Européenne de Sécurité des Aliments
AFSSA Agence Française de la Sécurité Sanitaire des Aliments
AFSSAPS Agence Française de Sécurité Sanitaire des Produits de Santé
AFSSE Agence Française de Sécurité Sanitaire Environnementale
AINS Anti-inflammatoires non stéroïdiens
AMM Autorisation de Mise sur le Marché
ANAES Agence Nationale d'Accréditation et d'Évaluation en Santé
ANMV Agence nationale du médicament vétérinaire
BRGM Bureau de recherches géologiques et minières
CDC Center for Disease Control and Prevention
CFES Comité Français d'Éducation pour la Santé
CGG Commission du génie génétique
CGB Commission du génie biomoléculaire
CIRAD Centre de coopération Internationale en Recherche Agronomique pour le Développement
CIRE Cellules interrégionales d'épidémiologie
CJCE Cour de Justice des Communautés Européennes
CNAMTS Caisse Nationale de l'Assurance Maladie des Travailleurs Salariés
CNEVA Ancien centre national d'études vétérinaires et alimentaires, intégré à l'AFSSA en 1998
CNRS Centre National de la Recherche Scientifique
CNSP Comité National de Santé Publique
CNSS Comité National de Sécurité Sanitaire
COGIC Comité de gestion interministérielle des crises
COMTOX Commission d'étude de la toxicité des produits antiparasitaires à usage agricole et des produits assimilés
COPERCI Comité permanent de coordination des inspections du ministère de l'Agriculture, de l'Alimentation, de la Pêche et des Affaires Rurales
CSHPF Conseil Supérieur d'Hygiène Publique de France
CSP Code de la Santé Publique
DDASS Direction Départementale des Affaires Sanitaires et Sociales
DERNS Direction de l'évaluation des risques nutritionnels et sanitaires
DGAL Direction Générale de l'Alimentation
DGAS Direction Générale de l'Action Sociale
DGCCRF Direction Générale de la Concurrence, de la Consommation et de la Répression des Fraudes
DGSanco Direction générale santé des consommateurs - Commission européenne
DHOS Direction de l'Hospitalisation et de l'Organisation des Soins
EFG Etablissement français des greffes
EFS Etablissement français du sang
EMEA European Agency of the evaluation of Medicinal Product - Agence européenne pour l'évaluation des médicaments
ESB Encéphalopathie spongiforme bovine
ESST Encéphalopathies subaigües spongiformes transmissibles
FAO Food and Agriculture Organisation - Organisation de l'alimentation et de l'agriculture (des Nations-Unies)
FDA Food and Drug Administration - Administration de l'alimentation et des médicaments (Etats-Unis)
FMC Formation médicale continue
FOPIM Fonds de promotion de l'information médicale et médico-économique
FSA Food Standards Agency - Agence de sécurité des aliments (Royaume-Uni)
HAS Haute Autorité de Santé
HCS Haut Conseil de la Santé
HCSP Haut Comité de la santé publique
IFEN Institut Français de l'Environnement
IGAS Inspection générale des affaires sociales
INERIS Institut National de l'Environnement Industriel et des Risques
INPES Institut National de Prévention et d'Éducation pour la Santé
INRA Institut National de la Recherche Agronomique
INSERM Institut National de la Santé et de la Recherche Médicale
InVS ou IVS Institut de Veille Sanitaire
IPSN Institut de Protection de Sûreté Nucléaire
IRSN Institut de Radioprotection et de Sûreté Nucléaire
JAMA Journal of American Medical Association
LEEM Les Entreprises du Médicament
LOLF Loi organique relative aux lois de finances
MHRA Medicine and Healthcare Products Regulatory Agency - Agence de réglementation des médicaments et des produits de santé (Royaume-Uni)
NIH National Institutes of Health - Instituts Nationaux de la Santé (Etats-Unis)
OGM Organisme génétiquement modifié
OMS Organisation Mondiale de la Santé
OPRI Office de Protection contre les Rayonnements Ionisants
RNSP Réseau National de Santé Publique
SRAS Syndrome Respiratoire Aigu Sévère
SSM Structure scientifique mixte (INRA - ministère de l'Agriculture)
THS Traitement hormonal substitutif
PRÉFACE
Des lois récentes ont redéfini le cadre de la sécurité sanitaire et environnementale, répondant aux attentes de la société et aux crises diverses qui ont pu survenir.
Ces lois, celle du 1er juillet 1998 comme celle du 9 mai 2001, ont précisé les modalités de la vérification de leur application. Dans ce cadre, l'Office Parlementaire d'Evaluation des Choix Scientifiques et Technologiques a été chargé par le législateur de procéder à l' « évaluation de l'application » de ces textes.
A cette fin, le 20 novembre 2002, l'Office a chargé M. Bernard SEILLIER, Sénateur de l'Aveyron, de conduire les travaux d'évaluation. Celui-ci a présenté le 29 janvier 2003 une communication sur le projet d'évaluation mais le rapport lui-même n'a pu être achevé avant le renouvellement de l'Office parlementaire d'évaluation des choix scientifiques et technologiques consécutif aux élections sénatoriales de septembre 2004. C'est dans ce contexte que l'Office m'a chargé de l'achèvement du rapport le 3 novembre 2004.
Cette situation particulière m'a amené à limiter le champ d'investigation du rapport sur l'application de la seule loi de 1998, l'application de la loi de 2001 créant l'AFSSE et l'IRSN devant être traitée ultérieurement par l'Office. La mise en place tardive et récente de ces deux instances justifie pleinement cette disjonction, la durée de leur fonctionnement ne permettant pas une analyse d'expérience suffisante. Cependant, le survol du dispositif global de sécurité sanitaire m'a conduit à aborder le positionnement de l'AFSSE et a formuler quelques recommandations qui peuvent éclairer dès maintenant les pouvoirs publics.
L'examen de l'AFSSA était largement engagé et ne soulevait pas de question particulière. La sécurité alimentaire est globalement consolidée par une agence qui a su s'installer et se faire reconnaître. Par contre, les travaux d'évaluation de l'AFSSAPS étaient à réaliser. Ils ont été menés sous l'éclairage brutal de la crise qui secoue le monde du médicament depuis octobre 2004 avec, en particulier, l'affaire du Vioxx. Cette conjonction m'a conduit à accorder une place particulière au dispositif lié aux médicaments.
Le cadre d'un simple rapport d'évaluation de l'application de la loi ne m'autorisait pas à pousser mes investigations, mais il m'a semblé indispensable d'alerter les pouvoirs publics sur l'enjeu majeur de la crise du médicament qui vient de s'ouvrir et de les inviter à s'emparer d'un dossier d'ampleur mondiale qui peut ouvrir une nouvelle crise de confiance entre le monde de la science et nos concitoyens.
Claude SAUNIER
Sénateur des Côtes d'Armor
Vice-Président de l'Office
INTRODUCTION GÉNÉRALE
La société moderne exprime une attente croissante de sécurité dans les domaines les plus variés. Cette attente est légitime dans la mesure où elle est la marque d'une conception du progrès mis au service de la population et non des seuls impératifs financiers.
La loi du 1 er juillet 1998 a créé une architecture institutionnelle et fonctionnelle novatrice qui constitue une nouvelle étape dans la réalisation de la sécurité sanitaire en France.
On ne partait certes pas du néant car bien des éléments avaient déjà été créés antérieurement au cours de la précédente décennie avec, par exemple, l'Agence du médicament. En outre, des dispositifs plus traditionnels constituaient déjà des structures répondant aux besoins les plus courants : les laboratoires de l'INRA, du CNEVA ou de la DGCCRF en attestent ; leurs contributions supportaient sans difficulté la comparaison avec ceux de pays européens comparables à la France. Mais le déficit conceptuel qui s'était fait jour progressivement ou à l'occasion de crises telles que l'ESB (la « vache folle ») ou l'hormone de croissance, sans oublier le cas très spécifique du sang contaminé, avait clairement montré la nécessité de construire cette nouvelle architecture sans pour autant ignorer les acquis des dispositifs anciens mais en imposant les principes qui caractérisent le mieux cette nouvelle architecture : séparation des acteurs de l'évaluation du risque et de sa gestion, indépendance de l'expertise, transparence des situations et des décisions, désignation d'objectifs précis à chacun des intervenants.
La loi du 1 er juillet 1998 avec les agences sanitaires qu'elle a créées et les mécanismes qu'elle a systématisés, a répondu à ces besoins clairement identifiés.
Cette législation novatrice a été élaborée à la suite de travaux approfondis menés au sein de la commission des affaires sociales du Sénat à partir de 1996 sous l'impulsion et la persévérance constante et éclairée de M. Claude Huriet, alors rapporteur au sein de cette commission de la mission d'information sur « les conditions du renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme en France » puis co-auteur et rapporteur de la proposition de loi qui en est issue, ainsi que de M. Charles Descours, Président de cette mission. Il convient de souligner la qualité des travaux de ces parlementaires.
Par son article 30, cette loi comporte une disposition prévoyant son évaluation par le Gouvernement et par l'Office Parlementaire d'Évaluation des Choix Scientifiques et Technologiques, préalablement à un nouvel examen dans un délai de cinq ans à compter de son entrée en vigueur. Il s'agit donc là d'une « saisine automatique » prévue par le législateur. A ce jour, la seule saisine de ce type qui ait eu lieu a été celle comprise dans la loi n° 94-654 du 29 juillet 1994 « relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal 1 ( * ) » désignée couramment sous le nom de « loi sur la bioéthique » ; elle était prévue (art. 21) exactement dans les mêmes termes que ceux retenus pour la saisine dont nous sommes chargés aujourd'hui.
Telle est l'origine du présent rapport.
On se trouve évidemment ici dans un domaine complètement différent donc dans des situations peu comparables à ce seul précédent. L'éclairage principalement lié à l'éthique, au développement de découvertes scientifiques et de possibilités technologiques qui caractérisaient les dispositions sur la bioéthique ne se retrouvent pas ici, où il s'agit d'évaluer des dispositions visant de nouveaux mécanismes et structures de la sécurité et de la veille sanitaire et ce, en parallèle avec l'évaluation propre dont le Gouvernement est chargé de son côté, ce qui est tout à fait inédit.
En outre, la loi n° 2001-398 du 9 mai 2001 créant l'Agence française de sécurité sanitaire environnementale d'une part, et l'IRSN (Institut de radioprotection et de sûreté nucléaire, par fusion de l'IPSN (Institut de protection et de sûreté nucléaire) et de l'OPRI (Office de protection contre les rayonnements ionisants) d'autre part, a également prévu, sur le même modèle, la saisine de l'Office parlementaire d'évaluation des choix scientifiques et technologiques.
S'agissant de l'IRSN, un rapport d'information de la commission des finances de l'Assemblée nationale qui faisait suite à un contrôle sur pièces et sur place a été publié le 29 avril 2004, M. Philippe Rouault, député, en étant l'auteur. Il y a été procédé à l'analyse de la mise en place de l'IRSN, qui a donné lieu à des appréciations critiques sur les difficultés administratives et budgétaires rencontrées ; sur ce dernier point, il indiquait notamment (Rapport d'information, page 23, n° 1580 XIIè législature) : « Non seulement l'IRSN, établissement public à caractère industriel et commercial, a été créé sans apport de fonds de roulement de la part du CEA dont l'Institut est issu pour l'essentiel, mais encore la dotation budgétaire complémentaire qui lui a été allouée pour faire face, sans perte de substance, à ses nouvelles obligations fiscales de droit commun a été au départ sous-évaluée ».
De son côté, dans un rapport très récent sur le démantèlement des installations nucléaires et la gestion des déchets radioactifs, la Cour des Comptes « a critiqué l'incapacité récurrente des pouvoirs publics à désigner les dirigeants de certains organismes en temps utile : à deux reprises, le conseil d'administration et le président de l'ANDRA n'ont pas été renouvelés (octobre 1997 à janvier 1999 ; depuis décembre 2003) ; créé en février 2002, l'IRSN ne sera doté d'un président et d'un directeur général qu'en janvier 2003 ; depuis septembre 2003, le CSSIN (Conseil Supérieur de la Sûreté de l'Information Nucléaire) ne se réunit plus faute de renouvellement du mandat de ses membres » . (Synthèse du rapport - janvier 2005).
Ces retards et insuffisances et donc l'ensemble de l'application de la loi du 9 mai 2001 pour ce qui concerne l'IRSN pourraient être examinés spécifiquement dans un rapport de l'Office.
La nature et le cadre particuliers de ces saisines ont amené à réaliser le présent rapport selon des modalités adaptées. Il ne saurait être question de se livrer aux travaux d'analyse qui ne sont pas dans la vocation de l'Office lequel n'en n'a d'ailleurs pas les moyens, travaux du type de ceux que réalise la Cour des comptes, qu'il s'agisse de rapports proprement dits ou d'audits. Il ne s'agit pas plus de refaire les travaux des commissions parlementaires (Finances, Affaires sociales) dans le cadre des lois de finances annuelles. Il ne s'agit pas davantage, et pour les mêmes raisons, de réaliser des missions du genre de celles dont les inspections générales ont été ou sont chargées pour le compte du Gouvernement.
C'est vers l'adéquation aux objectifs fixés des institutions et mécanismes nouveaux dans le domaine de la sécurité sanitaire que le rapport a été orienté et ce, dans la perspective qui avait été tracée lors de l'examen de la loi du 1 er juillet 1998 d'une part, mais aussi à la lumière des évolutions observées depuis qui révèlent souvent de très rapides changements voire des bouleversements à travers certains événements, qu'il s'agisse de la crise de la canicule ou du problème posé par les produits phytosanitaires, et, plus récemment, par la crise du médicament.
En effet, l'analyse des situations et de leurs évolutions dans ces domaines depuis près de six ans, révèle des changements considérables quant aux réalités concrètes, mais qui doivent être mis en perspective.
Dès lors, il est logique de constater que si les choix faits en 1998 se sont révélés adaptés et judicieux dans le cadre de l'analyse de l'époque, et que la mise en application s'est faite d'une manière satisfaisante pour l'essentiel, l'évolution de certaines situations amène à prendre en compte d'autres considérations. Certaines démarches doivent être soit complétées, soit, au contraire, remises en cause ; cette remise en cause devra intervenir notamment lorsque l'illusion du risque zéro finit par obséder les décideurs en raison de pressions médiatiques excessives alors même que l'on ignore des risques réels qui ne bénéficient pas de coup de projecteur. Ajoutons que des énergies sont parfois mobilisées en pure perte sur des questions dont la consistance même n'a jamais été avérée (cas emblématique des antennes-relais de téléphonie mobile) et que des cas limites entraînent des dispositions fondées sur des normes injustifiées au seul motif que des instruments de mesure encore plus fins existent. Une lecture juste, pertinente, anticipatrice et raisonnable du principe de précaution serait souvent opportune.
L'ambition contenue dans la loi de 1998 était donc légitime et a été concrétisée dans les textes puis sur le terrain de façon satisfaisante. En revanche, elle a pu donner l'illusion que la construction d'une architecture institutionnelle bien adaptée, l'existence de principes clairs et respectés et d'une transparence assurée permettraient de maîtriser tout type de situation. L'éclairage médiatique, nécessaire mais parfois réducteur, conduit à une déformation des proportions ; tout incident, toute intoxication sont d'emblée qualifiés de « crise sanitaire », même si on aperçoit assez rapidement que dans un cas de listeria la chaîne du froid n'a pas été respectée par les consommateurs pour un produit qui s'est révélé irréprochable, ce qui n'a pas empêché le fabricant de faire ensuite faillite. Les notions d'alerte, de « veille sanitaire », de crise, doivent être bien définies, mais le cahier des charges de chacun des acteurs, s'il doit être précis, ne peut pas se présenter comme un mécanisme d'horlogerie. La loi et le règlement, ne sauraient tout prévoir à tous les échelons à tout moment ; l'organisation anticipative poussée à l'excès à travers des programmes exclusifs de l'esprit de veille peut entraîner de graves erreurs de jugement comme on a pu le constater avec la canicule.
La FDA (Food and Drug Administration) américaine elle-même, qui est longtemps apparue comme la référence suprême, suscite aujourd'hui des incertitudes sur sa structure et son fonctionnement.
Les évolutions rapides des réalités concrètes tant technologiques que politiques, appellent également une remise en perspective des mécanismes et des objectifs en matière de sécurité sanitaire. Les risques nutritionnels ont pris une dimension nouvelle et quelquefois dramatique au cours des dernières années : en peu de temps, on est passé du niveau des simples retouches nécessaires dans un paysage plutôt progressif avec, par exemple, l'amélioration de la prévention générale des maladies cardio-vasculaires (graisses, sel, exercice) à une prise de conscience très brutale et récente de l'irruption de l'obésité, aux multiples conséquences, avec des comportements alimentaires régressifs.
Sous un autre angle, la mondialisation accélérée dans le domaine alimentaire exige de véritables prouesses si l'on veut que le haut niveau de sécurité atteint aujourd'hui se maintienne. La traçabilité pour la viande a fait des progrès considérables à la suite de la crise de la vache folle mais on voit tout de suite à quels nouveaux défis la sécurité alimentaire est confrontée avec la multiplication des échanges et ses conséquences pour la préparation de plats élaborés aux matières premières d'origine très diverses. La recherche fructueuse réalisée en urgence en mai 2003 par la DGCCRF et l'AFSSA à la recherche d'un colorant cancérogène dans un piment importé illustre ce type de défi. L'ouverture à dix nouveaux membres de l'Union européenne est en train de révéler certaines difficultés nouvelles dans ce même domaine alimentaire et dans celui du médicament.
***
L'ensemble de ces éclairages, administratifs et factuels, contribuent à l'évaluation des différents aspects du dispositif de sécurité sanitaire français et à sa mise en perspective par rapport aux objectifs affichés, aux besoins réels et aux exigences légitimes.
A travers une analyse générale, on notera les acquis indiscutables et nombreux que la législation de 1998 a apportés mais aussi les interrogations que sa mise en place suscite aujourd'hui où la tentation de tangenter le risque zéro pose de réels problèmes dans les secteurs les plus divers de la sécurité sanitaire.
On traitera ensuite plus particulièrement le domaine de la sécurité sanitaire des aliments en consacrant un développement particulier à l'AFSSA, y compris sous l'angle européen.
L'AFSSAPS, son fonctionnement et son domaine d'activité feront de la même manière l'objet d'un examen ; au-delà du fonctionnement courant, deux développements seront consacrés à des questions d'une intense actualité dans le domaine des produits de santé soulevés notamment par certains médicaments, et, en second lieu, dans l'apparition de nouvelles formes de dispensation de médicaments qui pourrait préfigurer des régressions à haut risque.
Une réflexion d'ensemble sur l'architecture institutionnelle générale, notamment le positionnement de l'AFSSE (loi du 9 mai 2001) constituera la quatrième partie de cette évaluation.
PREMIÈRE
PARTIE :
LA SÉCURITÉ SANITAIRE : ÉTAT DES
LIEUX
Un survol rapide mais global du concept de sécurité sanitaire fait immédiatement prendre conscience des progrès considérables qui ont été accomplis en France au cours des quinze dernières années, plus spécialement depuis la loi du 1 er juillet 1998 relative au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme.
On doit ces avancées à la pression de crises sanitaires réelles et d'alertes multiples, mais aussi à l'engagement de plusieurs catégories d'acteurs. Cet acquis doit être très brièvement rappelé pour indiquer dans quelles conditions il a déterminé la réalité institutionnelle et factuelle d'aujourd'hui.
En second lieu, l'acquis conduit à des interrogations lourdes sur la situation actuelle qui connut des réussites et des échecs : le drame de la canicule de 2003 est là pour appeler à la réflexion, constructive si possible.
I. L'ACQUIS
1.1. De la santé publique à la sécurité sanitaire
Le XIXè siècle
Les dispositions qui assurent l'hygiène publique dans un Etat moderne remontent fort loin dans le temps. Au-delà des déclarations de principe fixant des objectifs de salubrité publique aux autorités locales alors en cours d'institutionnalisation, une loi de 1822 posait des règles en matière d'épidémie. Les épidémies de choléra qui marqueront toute la première moitié du XIXè siècle attireront déjà l'attention d'une manière dramatique sur la nécessité de mesures urgentes en ce domaine. Ce fléau n'est pas exclusif de quelques autres, y compris, on l'a trop vite oublié, le paludisme qui sévit en Europe jusqu'à des périodes récentes.
Ces épidémies, souvent dramatiques, ont conduit à des initiatives de nature différente : ainsi, les travaux d'assèchement des marais ont constitué un impératif clairement identifié et mis en oeuvre. Une nouvelle épidémie de choléra qui toucha le département du Var en 1884 a largement contribué à la perception des besoins urgents dans ce domaine et à la nécessité d'une législation spécifique.
Le XXè siècle.
Le terme de santé publique est consacré par la loi du 15 février 1902 (relative à la protection de la santé publique). La place des autorités municipales et préfectorales y est clairement affirmée, notamment en cas d'épidémie. Elle impose, en outre, la vaccination obligatoire contre la variole ce qui constitue un pas considérable en matière de sécurité sanitaire. Elle s'impose aux droits personnels des citoyens et même au prix d'un risque dont on sait aujourd'hui avec certitude qu'il est très faible, mais réel. Comment cette mesure serait-elle aujourd'hui appréciée à l'aune des principes qui depuis sont ou se sont imposés en matière de risque ? La question est très actuelle. On rappellera sur ce point qu'il a fallu attendre le milieu des années soixante-dix pour qu'une loi impose la prise en charge par l'Etat des conséquences des accidents dus à une vaccination obligatoire.
C'est aussi à cette époque que le premier dispositif d'expertise sanitaire est créé avec le Comité consultatif d'hygiène publique de France. Fort peu de temps après, les bases juridiques modernes de la sécurité des aliments, plus particulièrement étaient fixées avec la loi de 1905 sur la répression des fraudes. L'outil essentiel de contrôle et de gestion du risque qu'est la DGCCRF trouve là ses origines.
Les développements scientifiques et technologiques ont certes bouleversé les conditions d'exercice de la médecine et de la production alimentaire, mais les bases de ces législations très considérablement étoffées et modernisées n'ont pas connu de véritable révolution jusqu'à la fin des années quatre-vingts.
Notons également qu'on n'a pas assisté à une judiciarisation alors que certains accidents ont donné lieu à des poursuites. Ainsi l'affaire du talc Morange qui fit une trentaine de morts et laissa de graves séquelles à près de deux cents autres personnes. Il est vrai aussi qu'avant de connaître la judiciarisation récente mais croissante de l'activité médicale qui peut confiner au harcèlement, on a connu fort longtemps une immunité de fait qui n'était pas davantage satisfaisante.
Les crises de la fin du XXè siècle
En France, à la fin des années quatre-vingts, c'est précisément par la tragédie du sang contaminé que le mouvement vers la sécurité sanitaire s'est engagé. Résumant simplement la situation d'ensemble, le rapport annuel de l'IGAS (inspection générale des affaires sociales) précisait en 1992 : « la santé publique : une organisation inadaptée ».
Dans le rapport d'information du Sénat en 1997 (n° 196) 2 ( * ) , MM. Claude Huriet et Charles Descours caractérisaient ainsi la situation de cette administration : « la pauvreté des moyens financiers et humains de l'administration sanitaire a certainement été à l'origine de bien des dysfonctionnements que l'on a reprochés au ministère de la santé alors qu'il n'en était pas vraiment responsable ».
Ils avançaient dans la nouvelle approche de la sécurité sanitaire plusieurs principes d'organisation, outre celui de suffisance des moyens financiers et humains :
le principe d'autonomie et de responsabilité :
« L'affaire du sang contaminé avait montré les limites d'une organisation sanitaire ministérielle au sein de laquelle la dilution des compétences et la multiplicité des intervenants faisaient que l'on ne pouvait véritablement identifier le lieu de décision et de responsabilité.
C'est pourquoi, lorsque l'Etat a choisi de réformer l'administration sanitaire au début des années 1990, il a choisi le statut d'établissement public pour l'Agence française du sang, l'Agence du médicament et l'Etablissement français des greffes. Ce statut, qui illustre le principe de la décentralisation fonctionnelle, permet de bien identifier l'autorité décisionnaire qui agit au nom de l'Etat. Au bout du compte, c'est cependant l'Etat qui est responsable et le ministre chargé de la santé demeure politiquement responsable ».
le principe de spécialité :
« Afin d'assurer la pertinence et la crédibilité de leurs décisions, les institutions chargées de promouvoir la sécurité sanitaire des biens de santé doivent agir en fonction des seules préoccupations sanitaires, à l'exclusion notamment de considérations économiques ».
Complétant cette formulation, les mêmes auteurs pointaient ce qui constituait encore un grave défaut dans l'organisation de l'époque (1996) : l'absence de séparation des contrôleurs et des gestionnaires pour l'Agence française du sang et pour l'Etablissement français des greffes.
Ces considérations visant exclusivement la sphère sanitaire et non alimentaire, il convient d'en garder la spécificité à l'esprit. Il en va de même pour la présentation et l'analyse de la sécurité sanitaire par M. Didier Tabuteau dans l'ouvrage qui porte précisément ce nom 3 ( * ) . Avant de présenter ce qu'il appelle les « quatre principes cardinaux » autour desquels s'est progressivement structurée la sécurité sanitaire (principe d'évaluation, de précaution, d'impartialité et de transparence), il indique :
« Il est en effet vite apparu que la sécurité sanitaire comme les autres sécurités, et notamment la sécurité extérieure, ne s'improvise pas. Elle se construit par étape et requiert une méthodologie commune. Chaque maillon du dispositif n'a de réelle efficacité que pour autant qu'il s'insère dans un système cohérent, complet et dans lequel les fonctions et responsabilités sont identifiées, rationalisées et assurées. Comme en matière de défense, les moyens sont nécessaires mais insuffisants s'ils ne sont pas au service d'une stratégie ».
Dans le domaine alimentaire, le rapport de sénatorial précité 4 ( * ) , Charles Descours dressait un tableau critique : « La crise de l'ESB a révélé le caractère imparfait des procédures tendant à garantir la sécurité sanitaire des produits alimentaires, tant en France qu'au niveau communautaire. (...)
Ainsi,les travaux menés par la mission ont permis de constater que deux conditions principales de la sécurité sanitaire des produits alimentaires n'étaient pas réunies : la connaissance des risques liés à l'alimentation est très insuffisante pour fonder une réglementation adéquate, et les autorités chargées du contrôle ne disposent pas d'une indépendance suffisante par rapport aux intérêts des producteurs ; leur approche est essentiellement tournée vers la santé animale, ce qui ne suffit pas à garantir la santé de l'homme ... ».
Pour la veille sanitaire (notant que le réseau national de santé publique constituait un progrès insuffisant), l'appréciation, sans être aussi sévère, était néanmoins critique : « dotée de moyens insuffisants, elle n'est pas assez coordonnée ni performante ». Il soulignait plus particulièrement la multiplicité des organismes assurant directement ou non des missions de veille sanitaire dans les termes suivants :
« Ces organismes sont de statuts divers, et ils sont rattachés à des ministères différents. L'audition de responsables de beaucoup d'entre eux a donné aux membres de la mission le sentiment que chacun faisait un bon travail, disposait de personnels compétents et oeuvrait dans l'intérêt commun. Mais ces auditions ont également montré l'extrême cloisonnement entre la plupart des organismes, dont chacun ignore l'activité des autres. Elles ont également montré leur isolement et la faiblesse des procédures d'alerte ».
Cette remarque tout à fait incontestable à l'époque peut être gardée comme repère car si la création et le développement des activités de l'InVS constituent une amélioration substantielle, la multiplicité des instances périphériques est bien réelle et les cloisonnements ont contribué à l'erreur d'appréciation sans précédent qu'a été la canicule 2003.
1.2. Les principes d'organisation de la sécurité sanitaire
La première vague de structuration des instruments de la sécurité sanitaire au début des années quatre-vingt-dix a déjà fixé des éléments nouveaux dans un domaine marqué par des conceptions traditionnelles et routinières, sans parler de l'insuffisance criante et chronique des moyens indispensables. L'Agence française du sang, l'Agence du Médicament, puis l'Etablissement français des greffes ont ainsi constitué une réelle novation qui a aussi été un ensemble de repères pour la suite.
Pour la deuxième vague, celle de la loi de 1998, les réflexions et les travaux précités ont amené les décideurs à faire preuve d'une beaucoup plus grande ambition, tant pour les structures que pour les objectifs assignés à cette sécurité sanitaire.
Peut-être est-il paradoxal d'aborder les structures avant les objectifs, mais ce sont souvent celles-ci qui d'abord focalisent le débat et l'orientation des choix. Les objectifs seront donc réévalués dans le cadre des interrogations qu'ils suscitent.
Des agences spécifiques à chaque domaine
Les réalités et l'expérience américaines à travers la F.D.A. (Food and Drug Administration) , le secteur alimentaire et pharmaceutique et les CDC ( Centers For Disease Control) pour le secteur de la santé constituaient des références pour tous ceux qui réfléchissaient sur ces sujets et qui voulaient jeter les bases d'une structuration adaptée aux problèmes actuels et aux défis de l'avenir. La question principale est celle du choix de l'agence unique comme l'est la F.D.A. (avec de sérieuses nuances quant à sa compétence exclusive dans le domaine alimentaire, cf. infra) ou de deux agences distinctes, parti qui a été retenu en France avec l'AFSSA et l'AFSSAPS. Il n'y a pas lieu aujourd'hui de regretter ce choix et on observe qu'aujourd'hui personne n'envisage sérieusement l'Agence unique. Les Américains eux-mêmes, à l'époque déjà, n'avaient pas que des certitudes dans ce domaine.
Un éventail de compétences adapté
-- La spécificité des domaines impliquait que l'éventail des compétences soit adapté au cas par cas et ne soit pas l'objet d'une règle uniforme. Concrètement pour l'AFSSA, il y a séparation entre l'évaluation du risque dont elle a la charge et la gestion du risque qui reste de la responsabilité des directions générales des trois ministères concernés. L'analyse détaillée et la justification de cette situation sont données dans la partie spécifique à l'AFSSA.
Pour l'AFSSAPS, il y a intégration de l'évaluation et de la gestion au sein de l'Agence. La justification de cette disposition est d'abord une raison de sécurité sanitaire : la centralisation au niveau de l'agence de compétences autrefois réparties au plan national et local y garantit que les informations sur les anomalies constatées pour les médicaments ou d'autres produits de santé remontent directement à l'Agence par le biais de la procédure du signalement. Elle permet à l'Agence d'évaluer, de décider et d'agir très vite : le renforcement des compétences de l'agence par la loi de 1998 et l'élargissement de ses missions à tout le champ les produits de santé répond à ce souci.
Les considérations d'ordre économique et technique sont également importantes. Les délais excessivement long pour les AMM (Autorisations de mise sur le marché) que l'on connaissait au début de l'Agence du Médicament ont pu être progressivement très fortement réduits (de 2 ans à 4 mois entre 1992 et 2002) grâce à une structuration plus efficace de l'évaluation par des comités d'experts étoffés. Si certains progrès restent à faire au niveau de la gestion des AMM, cela n'est pas comparable à ce qui a existé précédemment.
-- L'extension des compétences de l'AFSSAPS aux produits et dispositifs autres que les médicaments procède du constat peu rassurant fait en 1996-97 par le rapport précité (MM. Claude Huriet et Charles Descours). C'était particulièrement nécessaire pour les dispositifs médicaux où la procédure française était tout à fait déficiente et la procédure européenne nettement insuffisante. Les « produits frontières » ont également été inclus, donnant aussi à l'AFSSAPS les compétences nécessaires pour atteindre aussi l'objectif de sécurité sanitaire ; on a pu observer à plusieurs reprises tout l'intérêt de cette extension avec le cas des cosmétiques ou des produits pour les lentilles de contact par exemple.
1.3. Les instruments initiaux de la sécurité sanitaire
L'Institut de veille sanitaire (InVS), tête de réseau
? Ses premières responsabilités
-- La présentation de cette nouvelle structure contribuant à l'objectif primordial de sécurité sanitaire a été donnée en 1997 (rapport précité du Sénat n° 196, page 66), dans des termes qui méritent d'être rappelés tels quels car ils illustrent d'une manière concise et frappante l'inspiration du projet, sa portée et aussi, à l'expérience des événements, ses limites :
« Assurer la veille sanitaire en créant un Institut de la veille sanitaire
Au terme du recensement des divers organismes susceptibles d'avoir une activité de veille sanitaire, votre commission a constaté à la fois le foisonnement de telles structures, leur quasi-absence de coordination et l'inexistence d'un système d'alerte approprié qui pourrait permettre aux ministres responsables d'agir opportunément et sans délai.
Aussi propose-t-elle la constitution d'un Institut de veille sanitaire qui constituerait une tête de réseau pour la fonction de veille sanitaire, un peu à l'image de ce qui existe aux Etats-Unis avec les Centers for Disease Control.
L'institut de veille sanitaire aurait une triple mission de surveillance, d'étude et de recommandation.
Cet organisme serait obligatoirement destinataire de toutes les informations utiles collectées par les autres organismes qui doivent continuer d'exister, ne serait-ce que parce qu'ils remplissent le plus souvent d'autres missions que celle d'assurer la veille sanitaire. Il serait également destinataire d'informations non nominatives résultant de la transmission aux caisses de sécurité sociale de données issues du codage des actes et des prescriptions.
Ayant reçu ces informations, il pourrait en faire le tri et mener les enquêtes qu'elles justifient afin de détecter l'origine des événements constatés pour la santé de la population.
Les enseignements tirés de ces enquêtes feraient l'objet de recommandations aux pouvoirs publics : ils seraient transmis, pour décision, au comité permanent qui fait l'objet de la quatrième proposition ».
La loi fixe avec précision les mesures concrètes dont il est chargé et notamment celles qui lui étant spécialement attribuées visent l'alerte à travers la veille qui est sa raison d'être :
« Art. L. 1413-2 - Un Institut de veille sanitaire, établissement public de l'état, placé sous la tutelle du ministre chargé de la santé, est chargé :
1 ) D'effectuer la surveillance et l'observation permanente de l'état de santé de la population, en s'appuyant notamment sur ses correspondants publics et privés, participant à un réseau national de santé publique, dans le but :
- de participer au recueil et au traitement des données sur l'état de santé de la population à des fins épidémiologiques ;
- de rassembler, analyser et actualiser les connaissances sur les risques sanitaires, leurs causes et leurs évolutions ;
- de détecter tout événement modifiant ou susceptible d'altérer l'état de santé de la population ;
2°) D'alerter les pouvoirs publics, notamment l'Agence française de sécurité sanitaire des produits de santé mentionnée à l'article L. 5311-1, l'Agence française de sécurité sanitaire des aliments mentionnée à l'article L. 1323-1 et l'Agence française de sécurité sanitaire environnementale mentionnée à l'article L. 1335-3-1, en cas de menace pour la santé publique, quelle qu'en soit l'origine, et de leur recommander toute mesure ou action appropriée ;
3°) De mener à bien toute action nécessaire pour identifier les causes d'une modification de l'état de santé de la population, notamment en situation d'urgence.
On peut dire, à la lecture de ces dispositions (notamment « détection de tout événement modifiant ou susceptible d'altérer l'état de santé de la population (...), alerter les pouvoirs publics en cas de menace pour la santé publique, quelle qu'en soit l'origine ») et à la lumière des travaux préparatoires, que la volonté des législateurs a été globalement respectée.
? Ses développements ultérieurs
-- L'InVS a recueilli l'acquis du réseau national de santé publique et développé ses compétences dans ses nouveaux secteurs comme les maladies chroniques ou les investigations sur alerte.
Les moyens ont permis à l'InVS de s'installer et de s'étoffer. L'effectif au réseau national de santé public était de 70 personnes en 1998 ; les effectifs budgétaires autorisés sont passés de 100 en 1999 à 209 en 2002. Des difficultés liées à des statuts inexistants ou inadéquats ont pu freiner le recrutement effectif de personnel alors qu'on ne dispose en France de très peu de personnes de qualification satisfaisante dans l'épidémiologie d'intervention ou l'évaluation des risques sanitaires.
La conclusion du COM (contrats d'objectifs et de moyens) est intervenue en avril 2002 pour l'InVS après deux ans de préparation. Cette situation clarifiée par le COM est moins bonne pour l'AFFSAPS et l'AFSSA. Malgré de nombreuses déclarations d'intention et les échéanciers successifs, un tel contrat n'a toujours pas été conclu pour ces deux agences. La recherche de la responsabilité de ces retards ne nous incombe pas, mais le fait doit être souligné dans le cadre de l'évaluation de la loi de 1998, d'autant que la Cour des Comptes, se penchant sur cette question essentielle pour la modernisation des pratiques administratives, notait avec un optimisme qui paraît excessif aujourd'hui 5 ( * ) :
« La DGS a désormais mis au point une méthodologie d'élaboration de ces contrats d'objectifs et de moyens. Cette procédure type devrait faciliter leur généralisation aux autres agences sanitaires, en harmonisant les démarches et en définissant des critères d'analyse, de diagnostic et de suivi standardisés par des indicateurs. Cette démarche était d'autant plus nécessaire qu'elle constitue un élément du contrôle de gestion que le ministère en charge de la santé développe dans le cadre de la mise en oeuvre de la loi organique du 1 er août 2001 ».
Nous retiendrons l'absence de COM pour l'AFFSAPS et l'AFSSA comme une faiblesse réelle de l'application de la loi de 1998.
L'accomplissement des missions de l'InVS implique sur le plan local, outre les coordinations évidentes avec d'autres organismes (cf. infra), l'existence de correspondants. Il dispose à cette fin des CIRE (cellules interrégionales d'épidémiologie) et des réseaux régionaux dans le cadre de la santé au travail. Les CIRE, nées en 1995 ont été progressivement renforcées, mais devraient être encore étoffées pour pouvoir couvrir l'ensemble des thématiques traitées par l'InVS. Une illustration de leur intervention a été récemment fournie par la crise de la légionellose dans le Pas-de-Calais (hiver 2003).
Le domaine de la veille sanitaire est donc aussi réparti et organisé avec d'autres agences ou instances chargées d'une mission spécifique sur le terrain et qui permettent à l'InVS d'exercer précisément cette fonction de « tête de réseau ».
-- Ainsi, l'InVS est destinataire de tous les rapports relatifs à la veille sanitaire établis par l'AFFSAPS, l'AFSSA, l'EFG sur les produits dont elles ont la charge et sur ceux établis par tous les services de l'Etat et établissements publics rattachés.
-- L'AFFSAPS fait procéder à des enquêtes épidémiologiques sur les produits entrant dans le champ de ses compétences, en particulier sur les produits sanguins labiles et transmet les données épidémiologiques au ministre chargé de la santé et par extension à l'InVS ;
-- Dans le cadre de sa mission d'hémovigilance, l'EFS est ainsi tenu d'assurer la transmission des données relatives à la sécurité sanitaire des produits sanguins à l'AFFSAPS et les données épidémiologiques à l'InVS.
La fonction de tête de réseau de l'InVS est destinée à permettre la surveillance et l'alerte des autorités ministérielles et des autres agences en cas de menace pour la santé publique et la formulation des recommandations et des mesures en direction de ces agences. L'InVS bénéficie de domaines d'intervention complémentaires avec chacune d'entre elles, concernant notamment :
- les maladies infectieuses (commun avec l'AFFSAPS pour les risques concernant le SIDA,les hépatites virales, les maladies évitables par la vaccination, avec l'AFSSA pour les infections d'origine alimentaire, avec l'AFFSSAPS et l'AFSSA pour la maladie de Creutzfeldt-Jakob) ;
- les maladies chroniques (commun avec l'IRSN pour ce qui concerne la surveillance des cancers, avec l'AFSSA pour les risques liés à la nutrition) ;
- la santé environnementale (commun avec l'AFSSE et l'AFSSA pour les risques de pollution liés à l'eau et aux matières fertilisantes et supports de culture, avec l'IRSN pour les risques liés à l'exposition à des émissions radioactives).
L'InVS a précisément illustré avec soin les exemples de veille sanitaire qu'il mène en liaison étroite avec d'autres agences ou instances 6 ( * ) :
« L'AFSSA entretient des liens réguliers avec l'InVS. L'AFSSA et le DMI collaborent sur les points suivants :
- L'AFSSA apporte son expertise, sa contribution à la surveillance et à l'alerte puisqu'elle héberge deux CNR (Francisella tularensis, Brucella) et le laboratoire associé au CNR charbon de l'Institut Pasteur ;
- L'InVS contribue à l'expertise de l'AFSSA : participation au comité d'experts spécialisés « Microbiologie » et aux groupes de travail (expertises, E Coli VTEC, Listeria, Campylobacter, toxoplasmose, cryptosporidium ...) ;
- L'AFSSA participe en tant qu'expert à certains groupes de travail de l'InVS .
- plusieurs études sont réalisées en collaboration (diversité des souches de salmonelles, enquête « ferme » de l'étude SHU).
Par ailleurs, certaines saisines ministérielles sont conjointes. Il en est ainsi de l'étude de la morbidité et de la mortalité d'origine alimentaire où l'InVS a réalisé le travail pour les pathologies d'origine infectieuse en associant des experts de l'AFSSA, tandis que l'AFSSA traitait le volet toxicologique avec l'aide de certains experts de l'InVS.
L'enquête en population projetée pour explorer les habitudes nutritionnelles des français (Inca-2-PNNS) sera un autre exemple de collaboration, dans le cadre du programme national nutrition santé (PNNS).
- L'AFSSAPS collabore avec l'InVS sur des thèmes ciblés.
- L'InVS participe à l'hémovigilance en réalisant la surveillance épidémiologique des donneurs de sang, activité qui permet de mesurer les risques résiduels de transmission des principaux virus pathogènes transmis par le sang et de contribuer à l'évaluation de la sécurité transfusionnelle et des mesures de renforcement de la sécurité virale des produits sanguins.
Dans les investigations des infections nosocomiales, les liens avec la matériovigilance s'avèrent nécessaires. Ainsi, l'investigation de cas groupés d'hépatite C en institution a pu permettre d'incriminer des dispositifs médicaux (glucomètre par exemple) et le lien avec l'AFSSAPS a abouti à des recommandations sur leur usage. De même, les liens entre vigilance et surveillance dans le domaine de la vaccination se mettent en place. Dans certains cas, l'approche d'un problème complexe peut amener le ministère à saisir conjointement plusieurs agences pour y réfléchir. Tel a été le cas en 2002 pour aborder la question « aluminium et santé » où l'InVS a mené l'approche épidémiologique tandis que l'AFSSA approfondissait la question des apports alimentaires et l'AFSSAPS les apports médicamenteux et cosmétiques. Le tout devrait aboutir à une expertise globale inter-agences. Les rapports entre l'InVS et l'AFSSAPS sont régis par des comités de liaison annuels.
- L'AFSSE et l'InVS sont constamment en relation. La mise en place de cette nouvelle agence impose de préciser son champ de compétences et d'activités, en complémentarité de celui du département santé-environnement de l'InVS. Ceci conduit à de nombreux travaux conjoints notamment en ce qui concerne les rayonnements ionisants et non ionisants, la restructuration du réseau de toxicovigilance, ou la question de sécurité sanitaire relative aux incinérateurs d'ordures ménagères.
- L'INPES et l'InVS développent une complémentarité évidente.
L'InVS doit mettre à disposition de l'INPES tous les éléments épidémiologiques en sa possession pour lui permettre d'élaborer des programmes de prévention adaptés ».
(...)
Cet éventail de collaborations et d'actions conjointes est l'une des avancées les plus nettes que la loi de 1998 a permis. La progression est incontestable. De même l'efficacité avec laquelle certaines alertes sanitaires ont été traitées est à noter : SRAS (mars 2003), risque de grippe aviaire. Ces réussites, de longue haleine ou ponctuelles, ne sauraient faire oublier l'échec de taille qu'a été la canicule 2003 et qui met en cause, en outre, l'insuffisance ou l'excès ( ?) des instruments de coordination, au premier rang desquels le CNSS.
Le Comité National de la Sécurité Sanitaire (CNSS), instrument de coordination
Le CNSS (Comité National de la Sécurité Sanitaire) voulu en 1997 et prévu par la loi du 1 er juillet 1998, complétée par la loi du 9 mai 2001 devait donc être l'élément central de coordination de toutes les catégories d'acteurs. Le texte qui l'institue précise clairement son rôle à partir de ses tâches « d'analyser les événements susceptibles d'affecter la santé de la population », « d'assurer la coordination de la politique scientifique des agences » et de son obligation de se réunir « immédiatement en cas de déclenchement d'une crise sanitaire » :
« Art. L. 1413-1 - Un Comité national de la sécurité sanitaire est chargé d'analyser les événements susceptibles d'affecter la santé de la population, de confronter les informations disponibles et de s'assurer de la coordination des interventions des services de l'Etat et des établissements publics placés sous sa tutelle, notamment pour la gestion, le suivi et la communication des crises sanitaires. Ce comité s'assure également de la coordination de la politique scientifique de l'Institut de veille sanitaire, de l'Agence française de sécurité sanitaire des produits de santé, de l'Agence française de sécurité sanitaire des aliments et de l'Agence française de sécurité sanitaire environnementale.
Le Comité national de la sécurité sanitaire réunit, sous la présidence du ministre chargé de la santé, les directeurs généraux de l'Institut de veille sanitaire, de l'Agence française de sécurité sanitaire des produits de santé, de l'Agence française de sécurité sanitaire des aliments et de l'Agence française de sécurité sanitaire environnementale ainsi que les présidents des conseils scientifiques de ces trois agences et de l'Institut de veille sanitaire, une fois par trimestre, à la demande de l'un d'entre eux ou immédiatement en cas de déclenchement d'une crise sanitaire.
Il associe à ses travaux les autres ministres intéressés et notamment les ministres assurant la tutelle d'une agence. Il peut y associer toute autre personnalité ou organisme compétent ».
Cette lecture peut être complétée par un document de travail de la DGS d'avril 2002 7 ( * ) qui révèle une évolution de la conception du rôle de cette instance, par rapport à la loi et par rapport aux objectifs opérationnels, évolution sur laquelle on peut s'interroger :
« La démarche du CNSS est de constituer un fonds commun aux différentes parties sur l'analyse et la gestion des risques sanitaires.
Lors de ces séances, le Comité tire les conséquences des crises, évalue les risques émergents et tente d'élaborer une démarche innovante en matière d'analyse prospective et de veille scientifique.
Plus qu'une analyse à chaud des questions d'actualité, le CNSS privilégie le retour d'expérience. Au cours des différents CNSS, les sujets suivants ont été mis à l'ordre du jour, et certains ont fait l'objet d'un communiqué de presse : politique de lutte contre l'ESB, listériose, transfusion sanguine, les conséquences de la marée noire de l'Erika, aluminium et maladie d'Alzheimer, problèmes liés au piercing, éthers de glycol, dioxine, légionellose ...
Dans le même temps, trois groupes de travail ont été constitués au sein du CNSS sur les sujets suivants :
- critères, méthodes et procédures des processus de décision ;
- estimation quantitative du risque en situation d'incertitude ;
- analyse prospective des alertes ».
Le CNSS a visiblement été conçu pour faire face à des situations comme celles de la crise de l'ESB et par la nécessité d'établir une coordination à la fois dans l'action et dans l'analyse des risques sanitaires. Or, le texte de la DGS révèle un décalage qui l'a fait dévier vers des analyses de fond, vers des études qui d'ailleurs, pour certaines, n'ont pas été réalisées ou n'ont pas abouti : c'est le cas pour les deux derniers groupes de travail cités.
Quant à constituer l'échelon pour faire face aux crises sanitaires, le CNSS s'est révélé être un échec dont la DGS se doutait clairement dès 2002 à travers le texte précité : « Plus qu'une analyse à chaud des questions d'actualité, le CNSS privilégie le retour d'expérience ». Il est vrai qu'une instance de près de 70 personnes peut difficilement être efficace et opérationnelle dans une situation de crise.
Le CNSS n'a pas fait l'objet d'une demande de réunion exceptionnelle, que ce soit à la demande d'une des agences ou en raison d'une crise sanitaire. Il convient de noter d'ailleurs que la notion de « crise sanitaire » dont le déclenchement doit conduire aux termes de la loi du 9 mai 2001 à une réunion immédiate, n'avait pas fait l'objet d'une définition à la fin 2002. Il aurait peut-être fallu tirer alors la conclusion que cette instance ne pouvant remplir cette fonction, une autre aurait dû être désignée à cette fin, voire créé, avant que la canicule de 2003 révèle le vide organisationnel qui a contribué à l'aggravation de la crise avec l'absence totale de coordination de l'ensemble des acteurs du système de santé (santé publique, maisons de retraite, médecins de ville etc).
L'InVS lui-même, dans son rapport annuel (2002-septembre 2003, page 19) évoque ouvertement ce problème en pointant les retouches qui se profilaient dans le projet de loi relatif à la santé publique alors récemment déposé :
« La coordination de l'ensemble des agences sanitaires nationales est un objectif difficile à atteindre. Le CNSS en était l'instrument, mais ses modalités de fonctionnement amènent à réfléchir à des regroupements qui pourraient fédérer les agences d'expertise par produits par exemple, tandis que les structures transversales, InVS et Inpes, traitant respectivement de la surveillance de l'état de santé et de la préconisation des programmes de prévention qui s'imposent pour l'améliorer, pourraient être regroupés avec cohérence, comme c'est le cas dans d'autres pays. Si le projet de loi relatif à la santé publique ne prévoit pas ces regroupements, qui sont encore à l'étude, il remodèle déjà le CNSS en le fondant avec le Comité technique national de prévention pour devenir le Comité national de santé publique dont la vocation est la concertation et la coordination interministérielle des politiques de santé ».
La suppression du CNSS était devenue une nécessité évidente après la crise de la canicule.
La Direction Générale de la Santé (DGS), responsable de la Tutelle
Le corollaire de l'organisation de l'évaluation et éventuellement de la gestion des risques par des agences opérationnelles était naturellement le recentrage de l'activité du ministère de la santé, c'est-à-dire principalement la DGS autour de ses fonctions essentielles, c'est-à-dire la définition de la politique de santé, sa traduction normative et sa mise en oeuvre notamment en période de crise. C'est d'ailleurs ce que soulignait le rapport de MM. Claude Huriet et Charles Descours précité 8 ( * ) :
C'est pourquoi votre commission estime que l'administration centrale du ministère de la santé doit abandonner toutes les tâches qu'elle tente d'accomplir en doublon avec des organismes décentralisés, au profit de ses missions stratégiques : définition des choix de politique de santé, tutelle des organismes décentralisés, réglementation.
Un tel recentrage est indispensable si l'on veut que la santé de la population soit une priorité dans toutes les décisions de l'Etat : sécurité sanitaire des biens médicaux, mais aussi sécurité des biens de consommation, de l'alimentation, des milieux ».
Cette nécessité n'a pas été négligée tant dans l'allocation de moyens supplémentaires qui avaient été engagés de longue date qu'en ce qui concerne le recentrage des missions et de l'activité du ministère. Toutefois, des progrès sensibles restent à faire notamment dans l'exercice de la tutelle.
-- Les moyens humains de la DGS avaient augmenté avant même la création des agences. Ils avaient atteint un niveau en rapport avec les besoins d'une administration en charge de la politique de la santé à l'échelle de la France. Ils n'étaient plus dérisoires comme au début des années 90. A cet égard, on peut rappeler que la sous-direction chargée de l'ensemble de la veille sanitaire comptait quarante personnes. Ils ont encore été renforcés pour atteindre un niveau qui doit permettre de remplir un ensemble de missions recentrées. La DGS, par ce recentrage, dispose en fait de moyens nettement supérieurs, une part importante de tâches précédemment exercées par elle étant assurées par les agences. On verra par ailleurs (cf. infra) que celles-ci ont bénéficié de dotations importantes en personnel, ce qui était indispensable. A titre d'exemple, les effectifs de l'AFFSAPS représentent en 2003 cinq fois ceux de la direction de la pharmacie du ministère de la santé à la fin des années quatre-vingts.
-- la redéfinition du rôle de la DGS et notamment de l'exercice de la tutelle s'est concrétisée avec le décret n° 2000-685 qui fixe sa nouvelle structuration. Dans cette perspective a été créé un bureau spécifique « services déconcentrés et agences - SD4B » qui mobilise de façon transversale les sous-directions scientifiques et techniques. Il doit être rappelé qu'outre la DGS, d'autres directions participent également à la tutelle des agences, notamment la DAGPB.
-- L'exercice de la tutelle n'est pas aisé pour une administration face à une architecture comprenant autant d'agences autonomes, nouvelles pour la plupart. La création de la SD4B a été un progrès réel, mais la tutelle reste dans certains de ses aspects trop formaliste. Les COM (contrats d'objectifs et moyens) sont bien venus et souhaités, notamment par les agences qui n'en ont pas encore (AFFSAPS, AFSSA) ; ils peuvent toutefois constituer un élément d'ambiguïté dans la mesure où les agences pourraient par là même, chercher à stabiliser excessivement des objectifs et des éléments qui doivent rester adaptables aux évolutions, voire aux urgences 9 ( * ) . Cette remarque qui sera prolongée dans le développement suivant (« Les interrogations ») est également valable pour les travaux d'analyse et de programmation que la DGS mène, et en particulier ceux engagés en 2002-2003 dans le cadre de la préparation du projet de loi relatif à la santé publique. Le Pr Lucien Abenhaïm, directeur général de la santé, lors de son audition en novembre 2003 par la commission d'enquête de l'Assemblée nationale sur la canicule a lui-même déclaré à ce sujet 10 ( * ) :
« De combien de systèmes d'alerte peut-on disposer ? A-t-on essayé de réfléchir à l'avance ? J'ai ici un des six tomes de la consultation que j'ai organisée pour la première fois dans l'histoire de santé publique française à l'occasion de la préparation du projet de loi de santé publique en 2002 et 2003. Nous avons alors mobilisé non seulement une centaine d'experts, l'ensemble des agences de sécurité sanitaire, le haut comité de santé publique, les universitaires, mais également l'ensemble des acteurs de terrain dans les régions : médecins, représentants des usagers. Le but visait à identifier des objectifs de santé publique pour le pays.
Cent objectifs ont été identifiés, mais bien davantage ont été débattus. Sur l'ensemble de ces six tomes qui couvrent plusieurs milliers de pages, vous ne trouverez pas une seule fois le mot « canicule » ni l'expression « chaleur extrême ». Nous avons essayé de travailler et personne ne nous a signalé ce problème ».
Cette observation illustre on ne peut mieux le caractère particulièrement trompeur des assurances psychologique, administrative et politique que peuvent donner des programmations poussées à l'excès dans le domaine de la sécurité sanitaire. C'est une des interrogations que soulève le paysage actuel issu de la loi de 1998 et sa mise en oeuvre.
II. LES INTERROGATIONS
La démarche qui a crée un nouveau paysage de la sécurité sanitaire en France à partir de la loi du 1 er juillet 1998 a été validée par l'expérience. On observe à travers tous les témoignages et analyses une réelle et constante amélioration du niveau de sécurité atteint par les structures, les mécanismes, consolidant ce qui avait déjà été engagé, précédemment (Agence du médicament, comité d'experts spécialisés en matière alimentaire, réseau national de santé publique etc ...).
La remise en cause de l'architecture d'ensemble n'est donc pas à l'ordre du jour. Toutefois, les interrogations multiples illustrent la nécessité d'apprécier la portée des principes généraux sur lesquels cette réforme a été fondée et les aménagements et corrections que leur mise en oeuvre appelle. Au-delà des mécanismes de la loi de 1998, c'est l'ensemble des fondements de la sécurité sanitaire et environnementale qui sera abordé, les réalités concrètes constitueront des révélateurs efficaces des erreurs ou des insuffisances ou de développements nouveaux, source de risques.
Enfin, les éventuels réaménagements de l'ensemble des structures créées en 1998, et de leurs conditions de fonctionnement, élément obligé de la présente évaluation, seront traités pour autant que ces réaménagements paraissent nécessaires dans le développement consacré à la sécurité sanitaire des aliments pour l'AFSSA, les problèmes posés par l'architecture d'ensemble étant abordés dans la dernière partie du présent rapport (Une clarification nécessaire).
2.1. Des principes toujours globalement pertinents
Parmi les cinq principes présentés comme fondement de la nouvelle organisation de la sécurité et de la veille sanitaire, outre trois principes opératoires, deux principes généraux étaient retenus dans les termes suivants 11 ( * ) :
« 1. le risque zéro, qui ne peut être garanti, doit néanmoins être recherché par l'Etat, qui est garant de la sécurité sanitaire ;
2. Le principe de précaution doit toujours guider les autorités compétentes dans l'exercice de leur pouvoir de décision ».
On examinera successivement chacun de ces deux points.
2.1.1. La recherche du risque zéro
Le contexte historique (crise du sang contaminé et le risque de la transmission humaine de l'ESB principalement) dans lequel la nouvelle architecture de sécurité sanitaire a été conçue a naturellement renforcé les exigences dans ce domaine et partant, élevé le niveau des exigences.
Paradoxalement, les redressements sérieux qui avaient déjà été opérés à la fin de cette période, les améliorations constantes enregistrées dans des domaines moins sensibles ou moins médiatisés, ont donné l'impression que si le pire avait été frôlé, on pouvait maintenant espérer atteindre la meilleure sécurité et tangenter le risque zéro. En outre, le développement rapide des technologies très performantes semble donner la possibilité de réaliser cet objectif : la sécurité atteinte aujourd'hui en matière de produits sanguins pour la réduction du délai de latence pour la révélation d'un agent pathogène (VIH, prion pour la maladie de Creutzfeld-Jakob, hépatite C par exemple) est impressionnante.
Le recours au diagnostic génomique viral en 2001 a fait passer la probabilité du risque pour l'hépatite C de 1/800.000 à 1 pour 6 millions. Cette mesure a un coût de 21,3 millions d'euros par an. Cela montre d'ailleurs que la sécurité maximale a un coût et que la confrontation entre le prix de l'excellence et la limitation des moyens contraint les décideurs à des choix, toujours délicats.
Dans un domaine à l'inverse très peu marqué par la sophistication technologique, le drame de la canicule montre que si vraisemblablement le nombre de victimes aurait pu être limité en août 2003, il n'aurait certainement pas pu être réduit à zéro même en imaginant, hypothèse évidemment absurde, que chaque personne âgée à risque ait été reliée en permanence par un moyen électronique à un service de veille pouvant intervenir dans le délai exigé pour sa survie en cas d'hyperthermie maligne.
La tendance à croire le risque zéro accessible n'exclut d'ailleurs pas la recrudescence de comportements à risques, ce qui interpelle quelque peu les décideurs confrontés à des arbitrages délicats et devrait les conduire à consacrer plus d'énergie et de moyens aux grandes politiques de prévention.
Les perspectives tracées et les exigences fixées il y a maintenant plus de cinq ans ont permis plus qu'un simple progrès pour réaliser un saut qualitatif, mais ces ambitions légitimes ne sauraient reposer sur une prétention excessive qui peut être génératrice d'attentes illusoires.
2.1.2. Le principe de précaution
Jamais sans doute un principe de gouvernance et d'arbitrage au sein d'une société n'aura autant suscité de réflexions, discours, polémiques, que celui-là avec ce que connaît la France depuis plus de deux ans. On se limitera donc au simple rappel de son invocation dans la mesure où il a été utilisé comme principe d'orientation des décisions.
Un an après la mise en place des nouvelles agences, M. Claude Huriet 12 ( * ) évoquait précisément ce point en relativisant ou en tout cas en recadrant son affirmation précitée :
« Dans l'évolution du contexte national, je ne peux pas ne pas évoquer l'explosion du principe de précaution. C'est un sujet d'une actualité brûlante et peut-être ne serait-il pas souhaitable d'y consacrer trop de temps. Je voudrais souligner, dès le départ, qu'il y a pour nous une certaine contradiction entre les efforts accomplis par le législateur et les responsables de l'organisation nouvelle, et une application de plus en plus large du principe de précaution.
C'est pour le moment plus une réflexion qu'une interrogation, mais à quoi bon renforcer les dispositifs de sécurité de veille sanitaire si c'est pour amener les décideurs à appliquer le principe de précaution en oubliant un des critères qui était inscrit dans la loi Barnier de 1995, à savoir la proportionnalité de la réponse par rapport au risque réel ou supposé ? ».
Cette observation, bien antérieure au débat sur la Charte de l'environnement, mérite d'autant plus d'être soulignée.
L'invocation du principe de précaution devrait au moins être explicitée et cadrée dans les règles qui s'imposent à la mise en oeuvre (incertitude scientifique réelle, proportionnalité des mesures, réversibilité etc ...). Le recours implicite pour justifier une abstention surtout inspirée par le manque de lucidité, voire de courage, constitue une dérive particulièrement « polluante » dans le débat et la décision publique. La sécurité sanitaire est bien lointaine alors qu'elle seule devrait fonder l'application de ce principe. Pour peu que cette dernière soit liée au comportement désigné en anglais par l'acronyme « Nimby » (not in my backyard ; pas de ça chez moi), on aboutit à un dévoiement du principe dont l'invocation implicite ou explicite n'a plus beaucoup de rapport avec la philosophie d'origine ; les refus systématiques d'incinérateurs de déchets ménagers, notamment dans certains pays en Europe, corrélativement avec le refus d'assumer les conséquences de ce premier refus, sont une illustration de ce type de comportement.
L'exigence du respect des règles qui régissent la mise en oeuvre du principe de précaution s'impose à tous les intervenants et d'abord au décideur public. A ce titre, lorsqu'une analyse scientifique, a fortiori plusieurs, concluent à l'inexistence d'un risque, donc excluant la situation d'incertitude, il est regrettable que pour des raisons non explicitées mais que l'on pourrait qualifier de « psycho-politiques », une nouvelle expertise soit demandée ; à cet égard, la question des antennes relais de téléphonie mobile constitue un exemple concret de cette dérive. Immédiatement après une étude réalisée pendant l'année 2002 par l'Office parlementaire d'évaluation des choix scientifiques et technologiques, l'AFSSE avant même sa mise en place effective (décembre 2002), fut saisie en novembre 2002 d'une demande d'étude par la DGS. Ses conclusions, remises en juin 2003, concordants avec celles du rapport de MM. J.L. Lorrain et D. Raoul (sénateurs) pour l'Office, ne suffirent sans doute pas puisque l'AFSSE fut à nouveau saisie en septembre 2003 sur le même thème. La dépense d'autant d'énergie pour vérifier deux fois une absence de risque montre combien certaines pressions peuvent amener à un mauvais usage de principes sans précaution.
Le rapport de l'OPECST 13 ( * ) situait d'ailleurs clairement le problème dans ce cas en excluant précisément l'application du principe de précaution.
« Une situation paradoxale :
Les études scientifiques montrent clairement que s'il existe un risque lié à la téléphonie mobile, celui-ci est faible et a trait aux téléphones portables et non aux antennes relais. Aucune étude scientifique n'a en effet pu mettre en évidence des effets biologiques qui impliqueraient un risque sanitaire pour les populations vivant à proximité des stations de base de téléphonie mobile, compte tenu de leur faible niveau d'émission d'ondes électromagnétiques (en moyenne de l'ordre du dixième des valeurs de la recommandation européenne). Il convient de rappeler que la Tour Eiffel, avec ses émetteurs de télévision, représente une puissance analogue à celle de toutes les stations de base françaises réunies ».
Conclusion et recommandations :
Compte tenu des données scientifiques aujourd'hui disponibles à propos des effets de la téléphonie mobile sur la santé, il convient d'avoir recours à une attitude de prudence et de sagesse plutôt qu'au principe de précaution tel qu'il est notamment défini par la Commission européenne 14 ( * ) .
Ces mesures de prudence devront en tout état de cause être conformes aux principes généraux de bonne gestion des risques, c'est-à-dire :
-- proportionnées au niveau de protection recherche (et donc au niveau de risque à éviter) ;
-- cohérentes avec des mesures similaires déjà adoptées dans des domaines comparables ;
-- basées sur un examen des avantages et inconvénients de l'action ou de l'absence d'action ;
-- réexaminées périodiquement à la lumière des nouvelles données scientifiques ». (...)
Une autre conséquence éventuelle de l'invocation du principe de précaution ne doit pas être oubliée même si elle se pose plus sous l'angle de l'éthique sociale que de l'évaluation du dispositif de sécurité sanitaire : il s'agit de la mise en cause de la vaccination et naturellement de toute vaccination obligatoire. L'affaire, car il s'agit plus d'une campagne que d'une crise, du vaccin de l'hépatite B a montré comment cet acquis essentiel de santé publique pouvait être mis en cause par le moyen de doutes, certes recevables, mais qui ne justifiaient pas le dilemme dans lequel les autorités sanitaires ont alors été plongées. Qu'en sera-t-il le jour où l'on risquera dans le cadre du plan Biotox d'être en situation d'imposer à nouveau l'obligation de vaccination antivariolique, par exemple ?
Dans le domaine médical au sens large (pharmaceutique, etc) l'équation bénéfice/risque pour l'ensemble de la société reste le seul instrument de mesure et de décision, notamment lorsqu'il n'y a pas de doute scientifique quant à l'existence prouvée d'un risque. L'objectif de sécurité sanitaire collectif l'emporte sur la recherche de l'absence de risque individuel.
2.2. Des réalités qui imposent des réorientations
L'élévation constante du niveau de sécurité sanitaire dans les différents domaines abordés ici se confirme dans des proportions impressionnantes. Aux quelques exemples qui viennent d'être cités et ceux qui le seront dans les développements consacrés spécifiquement à la sécurité des aliments et à celle des produits de santé, on ajoutera ici quelques faits qui caractérisent la situation paradoxale que l'on connaît aujourd'hui : la confrontation entre un haut niveau de sécurité et une atmosphère d'inquiétude croissante.
Ce paradoxe conduit à durcir les normes. La justification de ce mouvement ne peut que susciter des interrogations dans des dispositifs où les repères quantitatifs ont une place essentielle dans les esprits. Mais leur validité sur le terrain est contestable, surtout lorsque certains d'entre eux font l'objet d'une instrumentalisation qui relève plus du fétichisme que de la réalité scientifique.
On observera d'ailleurs que la zone de progrès qui reste à couvrir en matière de sécurité sanitaire relève autant de l'usager que des instances responsables à condition que celles-ci mettent en oeuvre de véritables politiques de prévention fondées sur l'information des citoyens.
2.2.1 Un haut niveau de sécurité dans une atmosphère d'inquiétude croissante
Les progrès enregistrés d'une manière continue, mais encore plus nette depuis la réforme des structures de sécurité sanitaire, touchent tous les domaines.
Dans celui des produits sanguins , après le rétablissement urgent du début des années quatre-vingt-dix, on a noté (cf. supra) que des marges de sécurité substantielles ont encore été gagnées en ce qui concerne la réduction de la période de latence de révélation du risque. Ici comme ailleurs, les progrès scientifiques et technologiques en sont la cause directe, mais l'excellente organisation et gestion qui caractérise aujourd'hui l'EFS (Etablissement Français du Sang) y a également sa part et les réalités françaises dans ce secteur peuvent enfin constituer une référence à côté des pays étrangers les mieux placés.
Pour le médicament , le système français de l'AMM (autorisation de mise sur le marché) a gagné en rigueur, notamment à travers une expertise collective reconnue niveau communautaire, où l'AFSAPS et les spécialistes français travaillent directement pour l'Agence européenne du médicament et participent largement aux activités dans ce domaine. Le fait que la France n'ait pas connu du tout ou dans les mêmes proportions certains accidents enregistrés aux Etats-Unis avec un anticholestérique, la cérivastatine en 2001 et certains traitements hormonaux substitutifs après la ménopause pourrait être l'indice d'une situation satisfaisante. Mais au-delà de controverses naturelles au sein de la communauté scientifique, la prudence de l'AFSSAPS n'est jugée excessive qu'avant l'accident ...
Pour les dispositifs médicaux (expression qui recouvre de très nombreux « objets » depuis les pansements et literies d'hôpital jusqu'aux prothèses, orthèses vasculaires par exemple, lentilles optiques avec leurs produits d'entretien), les progrès ont été considérables ; les exemples qui viennent d'être cités ont tous de manière diverse et quelquefois en urgence, été l'objet de cette « matériovigilance ». La description de la situation en 1996 était particulièrement peu rassurante. La tâche pour l'AFSSAPS était d'autant plus lourde que sa compétence dans ce domaine était nouvelle et qu'elle n'héritait pas des moyens, de la structure et des pouvoirs d'une entité préexistante comme pour le médicament.
Dans le domaine de la veille sanitaire , de nombreux éléments positifs, que le drame de la canicule ne doit pas faire oublier, ont été enregistrés. On s'est habitué depuis longtemps déjà à la surveillance de la grippe et la production de vaccins ; cette réussite est le fruit d'efforts constants avec de multiples partenaires. On peut noter au passage que le recours aux médecins de ville, élément essentiel à l'efficacité du dispositif (réseau « sentinelle »), constitue un trop rare exemple de leur intégration dans les mécanismes de veille sanitaire.
Dans une toute autre problématique le risque constitué par la pneumopathie atypique dite SRAS à la fin de l'hiver 2003 a été traité dans des conditions de rigueur et d'efficacité qui traduisent une avancée indiscutable. Dans cet épisode, l'InVS a mis en ligne vingt personnes à temps plein pendant trois mois et renforcé les astreintes de nuit et de jours fériés. Il est peu probable que la précédente structure des instances compétentes aurait permis un tel fonctionnement. D'une manière évidemment moins visible, l'engagement des différentes parties prenantes au premier rang desquelles l'InVS dans le plan BIOTOX doit être souligné.
S'agissant de la sécurité sanitaire dans le domaine alimentaire , la progression est particulièrement mesurable, notamment par la survenance d'alertes successives sur le même risque comme la listeria. L'amélioration des mécanismes administratifs avec la création de l'AFSSA est évidente. L'efficacité de l'administration de tutelle chargée de la gestion du risque, la DGCCRF (Direction Générale de la Concurrence, de la Consommation et de la Répression des Fraudes) s'observe également illustrant ainsi cette réactivité, marque indiscutable d'un nouveau progrès. Ainsi, en mai 2003, cette administration, avertie de la présence sur le marché de piments colorés à l'aide d'un produit interdit B et fortement cancérogène, saisit l'AFSSA sous les 48 heures. Celle-ci rend dans le même délai un avis recommandant que les consommateurs ne soient pas exposés à ce produit. Le même jour, un plan de retrait du marché a été mis en oeuvre et le réseau d'alerte communautaire informé. La DGCCRF a également mis en place, à partir du 14 mai, un plan de contrôle de tous les lots qui avaient été vendus par ce fournisseur.
Alors qu'il s'agissait d'un produit importé de fort loin sur lequel on ne disposait pas d'éléments d'information aisément accessibles, cet exemple montre que le partage, dans le domaine alimentaire, entre l'évaluation et la gestion du risque n'interdit pas l'efficacité opérationnelle.
D'une manière générale, au-delà d'exemples tels que celui-ci, la réalité de l'élévation du niveau de la sécurité sanitaire dans le domaine alimentaire se vérifie à travers les résultats du plan de contrôle 2002 à la DGAL (direction générale de l'alimentation) sur les filières animales. Pour la viande de boucherie, en ce qui concerne les résidus chimiques (organophosphorés et PCB, organochlorés), on ne trouve qu'un cas positif sur 1684 prélèvements. Le résultat est du même ordre pour les antibiotiques et les stéroïdes. Dans la filière poulets, aucune trace de ce type de résidus n'a été trouvée. Dans le lait de vache, deux composants chimiques ont été trouvés, mais avec une occurrence très faible (entre 0,3 % et 1 %).
Les producteurs de la filière agro-alimentaire eux-mêmes, contribuent largement à cette progression en consacrant plus d'efforts encore à cet impératif de sécurité en terme d'hommes et de moyens techniques. Sur ce dernier point, la traçabilité électronique permet de retrouver et de rappeler les lots défectueux dans un délai de quelques heures, ce qui était impensable il y a encore quelques années. La distribution elle-même, comme la production, s'appuyant entre autres sur les normes HACCP (hazard analysis and critical control point) prolonge les actions menées par les administrations. La complémentarité des efforts des agents économiques rejoint et renforce la vigilance nécessaire des pouvoirs publics.
Dans le segment particulier que sont les marchés publics forains, où des lacunes notables perduraient, la mise en oeuvre de dispositions, en partie d'origine communautaire, a permis, avec l'aide des collectivités locales, d'atteindre un niveau de sécurité appréciable en un temps relativement bref.
Au-delà d'épisodes exemplaires et de considérations factuelles sur les facteurs de sécurisation, la mesure de la sécurité alimentaire s'apprécie en priorité à travers l'évaluation des accidents, donc des intoxications alimentaires. Cet aspect statistique n'est pas toujours aisé à cerner avec exactitude. Des éléments très concrets permettant une approche rigoureuse de la situation récente et de l'apprécier par des comparaisons dans l'espace et dans le temps, viennent d'être fournis par une étude approfondie réalisée conjointement par l'InVS et l'AFSSA publiée le 10 mai 2004. En voici les extraits les plus significatifs 15 ( * ) :
« Une analyse approfondie des données disponibles a permis à l'Institut de veille sanitaire d'estimer à plus de 200.000 (entre 238.836 et 269.085) le nombre moyen annuel de personnes atteintes de maladies d'origine alimentaire en France, au cours des années 1990. Les salmonelloses en sont la première cause (30 598 à 41 139 cas confirmés par an), suivies par les infections à campylobacter ».
« Principaux résultats de l'étude
Le nombre total annuel de cas hospitalisés pour une infection d'origine alimentaire a été estimé entre 10 188 et 17 771. Les salmonelloses en sont la première cause (5691 à 10 202 cas), suivies par les infections à campylobacter (2598 à 3516 cas) et la listériose (304 cas). La toxoplasmose apparaît comme la principale cause d'hospitalisation (426 cas) parmi les infections parasitaires étudiées.
L'estimation du nombre annuel total de décès se situe entre 228 et 691. Les infections bactériennes sont responsables de la majorité (84 % à 94 %) de ces décès avec une estimation de 191 à 652 décès annuels dont 92 à 535 liés aux salmonelloses, première cause de décès et 78 liés à la listériose, deuxième cause de décès.
La fréquence des maladies infectieuses d'origine alimentaire estimée dans notre étude apparaît très inférieure à celle estimée aux Etats-Unis (76 millions de cas) et en Grande-Bretagne (2 365 909 cas en 1995 en Angleterre et au Pays de Galles). Les effectifs estimés ne sont cependant pas strictement comparables. En effet, en Grande-Bretagne et aux Etats-Unis, les estimations portent sur les cas malades dans la population. De plus, la liste des agents pathogènes étudiés n'est pas identique et sont également incluses les gastro-entérites sans agent infectieux identifié qui ont un poids très important dans les estimations. Cependant, si les valeurs diffèrent sensiblement pour les raisons indiquées ci-dessus, les agents pathogènes responsables des plus grands nombres de cas, d'hospitalisations et de décès sont similaires, bien que les rangs soient différents ».
Il y a lieu de rappeler qu'en France, en 1950 le nombre annuel de décès dus à des intoxications alimentaires était estimé à 15.000. Il est maintenant estimé à un chiffre se situant dans une fourchette de 228 à 691 ; Aux Etats-Unis, il est actuellement évalué à 5000 par an. Ce nombre qui reste élevé 16 ( * ) car il constitue une amélioration par rapport à la situation connue il y a 10 ans (10.000) peut s'expliquer par, en premier lieu, une grande faiblesse des contrôles et paradoxalement une recherche excessive de l'absence de bactéries, ce qui permet un développement foudroyant des bactéries pathogènes lorsqu'une faiblesse intervient. Il convient de souligner en dernier lieu, l'inadaptation des comportements des consommateurs en aval du circuit alimentaire. Il illustre le problème des comportements à risque alors que l'on exige volontiers des distributeurs et des pouvoirs publics le « risque zéro ».
2.2.2. Les comportements à risque
Les comportements à risque évoqués au sujet de l'importance du nombre d'intoxications alimentaires aux Etats-Unis sont principalement relatif au non-respect de la chaîne du froid, ce qui peut également paraître paradoxal dans un pays où le réfrigérateur est l'élément mobilier le plus important de la cuisine, dans tous les sens du terme. Mais il semble que la phase de transport depuis le supermarché soit négligée dans l'organisation et ce, dans un climat comportant de forts épisodes de chaleurs extrêmes qui exige, sur presque tout le territoire national, des précautions particulières.
L'usage du réfrigérateur lui-même doit aussi être pris en considération et sur ce point qui fait souvent l'objet d'enquêtes, le comportement des Français mérite d'être évoqué car c'est là, il faut le rappeler, un chaînon essentiel qui reste généralement oublié.
Le CREDOC (Centre de Recherche pour l'Etude et l'Observation des Conditions de vie) à travers son enquête individuelle nationale sur la consommation alimentaire (INCA) réalisée en 1998-99 a fait une étude dont l'objectif était de connaître le niveau des températures de conservation et d'identifier les facteurs influençant le niveau des températures de l'air dans les réfrigérateurs.
Les conclusions ne sont évidemment pas très rassurantes :
« Sur le comportement des utilisateurs, on constate une méconnaissance du mode de réfrigération de leur réfrigérateur (36 % n'en ont aucune idée). D'autre part, les trois quarts environ des ménages règlent le thermostat de leur réfrigérateur dans les positions qui correspondent aux positions les moins froides, peut-être par souci d'économie d'énergie ou simplement pour un confort personnel pour certains aliments (boissons pas trop froide par exemple) en oubliant de se poser la question des risques hygiéniques éventuels.
Vingt-six pour cent des réfrigérateurs présentent une température globale supérieure à 8°C. Cette température est pourtant la température maximale tolérée pour les aliments dans l'arrêté réglementant l'hygiène des aliments remis directement au consommateur. De même, seuls 11 % des réfrigérateurs présentent une température inférieure ou égale à + 4 % C, qui est la température maximale tolérée pour de nombreux aliments tels que les produits transformés non stables à base de viande, comme les plats préparés réfrigérés ».
En outre dans 18 % des cas, la température dépassait 10°C. Enfin, l'entretient est un autre point faible : seul un tiers des appareils fait l'objet d'un nettoyage au moins une fois par trimestre. Des informations récentes confirment ces résultats : seules 5,6 % des viandes et 37 % des yaourts sont conservés à une température satisfaisante.
Le développement de certains modes peut contribuer à faire paraître ou réapparaître des risques, comme la vente en vrac.
Dans le domaine du médicament, bien des habitudes dangereuses se sont développées avec l'abus de l'auto-médication ou la gestion très personnelle des prescriptions. On observera (cf. infra AFSAPS) que nombre d'accidents où des médicaments sont en cause constituent un vrai problème de sécurité sanitaire (ce que recouvre l'expression « risque iatrogène »). En outre, le recours à des circuits ouvertement illégaux avec l'achat de médicaments par internet illustre l'actualité et la réalité de ce risque. (cf. infra AFSSAPS « Les risques émergents »).
Dans une perspective purement quantitative mais évidemment essentielle, on rappellera pour mémoire le choc entre le niveau de sécurité atteint pour les produits sanguins, alimentaires, pharmaceutiques, y compris en terme de coût marginal et les niveaux de risque actuellement atteints en matière d'obésité, d'alcoolisme, de toxicomanie et de tabagisme au niveau individuel et social.
2.2.3. La sécurité croissante vue à la loupe
L'amélioration considérable de la sécurité alimentaire par exemple résulte de la convergence de tous les facteurs ou acteurs évoqués : progrès scientifique et technique, producteurs et distributeurs, structures administratives, notamment d'évaluation et de contrôle, associations de consommateurs, moyens de la mesure et de la détection. Les progrès ont été considérables. Au-delà de l'alimentaire, cela se vérifie sous l'angle environnemental. Ainsi on ne recherche la présence de dioxines dans les aliments que depuis une douzaine d'années ; on en découvre naturellement des résidus alors que le risque moyen d'exposition a baissé de 50 % en quinze ans. L'efficacité et l'universalité des contrôles couplés à un fantastique progrès technologique ont donc un effet pervers dont il est temps de prendre conscience pour faire prévaloir une approche raisonnée de la sécurité sanitaire réelle. Sinon, moins il y a d'incidents et plus la loupe de la mesure sophistiquée conduit au développement de phobies en dehors de toute analyse objective de risque.
2.2.4. La sévérisation des normes : de la rigueur au fétichisme
Une norme fixant un seuil limite de présence d'une substance dangereuse ou supposée telle constitue le type d'élément concret, physique qui, s'il correspond aux résultats de travaux de recherche et d'expertise menés dans les règles de l'art, permet de fixer des règles objectives, intangibles, universelles et dont l'application est aisément contrôlable. On estime alors avoir fait tout ce qui est possible pour obtenir d'une part une législation pleinement satisfaisante au regard de la sécurité sanitaire et d'autre part une application opérationnelle.
Cette considération générale n'est pas contestable et est évidemment vérifiée quotidiennement et ce, dans tous les domaines. La connaissance qu'on a aujourd'hui de ses propres résultats d'analyses biologiques, le taux de cholestérol en étant un bon exemple, montre combien les éléments quantifiés parlent à chacun et le corps médical est lui-même confronté depuis longtemps de la part des patients à des « demandes d'analyses » fondées ou non.
Mais toute norme, comme « le » taux de cholestérol par exemple, n'est pas nécessairement suffisante et en tout cas doit être remise en perspective pour constituer la référence indiscutable d'un raisonnement scientifique valable, puis d'une action. Cette action doit elle-même être fondée par rapport à l'ensemble des objectifs fixés au-delà du but premier ; qu'il s'agisse de médecine ou d'environnement, on ne saurait se limiter au respect d'une seule norme en ignorant les « conséquences collatérales ».
Par ailleurs, et cela a déjà été évoqué, les progrès considérables des technologies de mesure ne sauraient faire perdre la raison en se fixant comme nouvel objectif la recherche de traces de traces. Le normes européenne de LMR (limite maximale de résidus) en vigueur depuis le 1 er juillet 2002 pour les aliments pour jeunes enfants a été fixée à 0,01 mg/kg ; elle est d'autant plus discutée qu'elle correspond en fait à la limite technologique de détection dans 40 % à 60 % des cas. L'atteinte de cette véritable limite ultime de la métrologie en matière alimentaire est développée dans un récent rapport de l'Office parlementaire d'évaluation des choix scientifiques et technologiques (« Les nouveaux apports de la science et de la technologie à la qualité et à la sécurité des aliments » page 89).
Les dérives auxquelles on assiste depuis plusieurs années se concrétisent généralement par un mouvement de durcissement successif de normes et aussi par la « diabolisation » d'éléments qui peuvent être en soi dangereux à un taux déterminé et dans un contexte multifactoriel. Ce qu'on relève également dans tous les cas de figure, c'est que lorsque l'échange d'arguments révèle une fragilité des partisans du durcissement continu des normes, ceux-ci finissent pas invoquer le caractère symbolique de la norme, ce qui autorise à parler véritablement de fétichisme : « La norme de 50mg/l de nitrate dans l'eau est politique », on ne saurait tenter de la remettre en cause. Dans de telles démarches, le progrès scientifique et technologique est enserré dans des oeillères particulièrement contraignantes : les découvertes sur les substances faisant l'objet de normes ne sont envisagées que dans un seul sens.
Les quelques exemples qui constituent, parmi d'autres, une illustration de ces dérives portent sur les nitrates, la norme de résidus en eaux profondes, les dioxines et le plomb dans l'eau.
2.2.4.1. Les nitrates
Elément naturel dont la concentration a des effets sur l'environnement et peut comporter des risques pour l'homme, les nitrates font l'objet d'une norme en ce qui concerne l'eau destinée à la consommation humaine : la concentration maximale pour qu'une eau soit potable est fixée à 50 mg/litre par la réglementation européenne, elle-même dérivée en fait de la norme OMS-FAO. Or, cette norme a une histoire particulièrement illustrative.
La survenance d'un nombre élevé d'une cyanose, précisément la « méthémoglobinémie », au nord des Etats-Unis à partir de 1945 et pendant les années cinquante entraîne la mise en lumière d'une concentration très élevée de nitrates dans l'eau qui était alors utilisée, notamment pour la préparation des biberons. On sait que pour la consommation humaine, ce ne sont pas directement les nitrates qui comportent un danger, mais un de leur sous-produits : les nitrites. En fait, on s'aperçut ultérieurement que ces eaux (puits en zones rurales le plus souvent) étaient tout simplement aussi contaminées par des bactéries pathogènes qui ont été alors mises en cause. Mais les certitudes énoncées sur le danger d'une certaine concentration de nitrates dans l'eau entraîna la fixation par un comité d'experts conjoints OMS-FAO de la dja (dose journalière admissible à 5 mg par kilo de poids corporel, soit 350 mg pour une personne de 70 kg. Faisant entrer la quantité de nitrate issue de la consommation de légumes, on fixa le chiffre à 100 mg/litre qui fut ensuite divisé par deux pour tenir compte des risques spécifiques encourus par les femmes enceintes et les nourrissons.
Il y a lieu de noter au passage que les légumes recèlent beaucoup plus de nitrates : au moins 2000 mg/kg dans la laitue, les navets et les épinards, sans oublier les carottes qui ne font l'objet d'aucune norme ni même mise en garde ou information et dont la consommation est même vivement recommandée. L'hypothèse scientifique formulée il y a plus de quarante ans s'est donc révélée fausse et les travaux américains récents (1999) ont définitivement disculpé les nitrates des accidents qui leur étaient précédemment imputés. Leur concentration à d'autres doses plus élevées mérite d'être surveillée, y compris pour des raisons environnementales, mais la « norme fétiche » de 50 mg/litre pour l'eau potable n'a pas, du stricte point de vue de la santé humaine, le fondement qu'on lui attribue. Le comité scientifique de l'alimentation humaine avait déjà indiqué, sur les nitrates, dans un avis le 19 octobre 1990 : « Le nitrate per se a une toxicité aiguë très faible et les effets nocifs rapportés résultent de la réduction en nitrites, soit avant ingestion, soit in vivo ». Mais le Pr Marion Apfelbaum, professeur de nutrition à la faculté de médecine de Xavier-Bichat (Paris) tire de cette observation la constatation suivante 17 ( * ) :
« Les deux seuils - 5 mg par kilo de poids corporels et 50 mg par litre d'eau de boisson - semblent animés d'une vie sociale propre. Les données récentes sur l'innocuité du nitrate chez l'homme et l'animal d'expérience n'y changent rien.
Pourquoi ? Les experts constituant les comités sont à l'évidence parfaitement informés. Et ils ne conseillent pas de supprimer la dose journalière admissible et le seuil de potabilité de 50 mg qui en découle, parce qu'ils ne peuvent le faire. Imaginons que, demain, ils annoncent que « l'eau est potable quelle que soit la concentration des nitrates qu'elle contient » et encore « qu'une feuille de laitue de 25 g contient autant de nitrate qu'un litre d'eau prétendument dangereuse ». (...)
Donc il faut persévérer dans l'être et continuer à faire comme si l'eau contenant plus de 50 mg par litre était à peine potable, et celle à plus de 100 mg par litre pas potable du tout. Même si le contraire est scientifiquement démontré.
Mais, puisque tout est ainsi pour le mieux dans le meilleur des mondes possibles, pourquoi en parler ? Car le dossier des nitrates est exemplaire de ce que la prétention de la science à dire le vrai universel est de plus en plus contestée, entre autres à cause de l'évidente symbiose du scientifique et du politique et de ce qu'un fait social puisse perdurer au-delà de ses causes ».
2.2.4.2. Les résidus en eaux profondes
La norme limitant la concentration maximum de pesticides dans les eaux profondes a été fixée il y a une quinzaine d'années à 0,1 g/litre. D'après les instances compétentes, elle n'a pas de fondement scientifique sauf pour ceux des produits dont la toxicité a été démontrée, auquel cas elle risque d'être insuffisamment sévère.
Dans le rapport de l'Office parlementaire d'évaluation des choix scientifiques et technologiques sur la qualité de l'eau et de l'assainissement en France, M. Gérard Miquel, Sénateur et rapporteur de l'étude, a rappelé l'ensemble des raisons qui mettent en cause la validité de cette norme :
« La fixation d'un seuil unique de pesticides dans l'eau fait l'objet de nombreuses controverses.
- La première critique porte sur le seuil unique. Il est observé que ce choix du seuil unique est un choix européen qui n'a pratiquement aucun équivalent au monde. Pour les pesticides, l'OMS a déterminé 40 valeurs guides (VG) différentes, adaptées aux différentes molécules.
- La deuxième critique porte sur le niveau choisi, beaucoup plus strict que les valeurs internationales et que les niveaux retenus par d'autres compétiteurs, notamment américains. Le rapport entre les valeurs limites européennes et les valeurs guides internationales peut varier de 1 à 3000 (pour le bentazone, la VG est de 300 g/l). L'Agence américaine de Protection de l'Environnement a fixé le seuil de l'alachlore, de l'atrazine et de la simazine, trois herbicides, à respectivement 2 g/l, 3 g/l et 17 g/l, soit un niveau de 20 à 170 fois plus élevé que la norme européenne.
La troisième critique porte sur une certaine incohérence dans la détermination des seuils. Tandis que l'attention était focalisée sur l'eau, les limites de résidus sur les produits d'alimentation traités aux pesticides n'ont pas été modifiées. On relèvera par exemple que les limites de résidus sur les fruits peuvent être jusqu'à 100.000 fois plus importantes que les teneurs acceptées dans l'eau. Cette situation suggère une sévérité excessive sur l'eau et que les normes appliquées à l'eau n'ont pas été fondées sur des raisons sanitaires.
La dernière critique porte sur une situation de blocage. On rappellera que les seuils actuels ont été fixés initialement il y a 25 ans. Pour le professeur Hartemann de la faculté de Nancy, « on pouvait fixer une norme de 0,1 g/l, par précaution, quand les connaissances scientifiques étaient encore limitées, mais à partir du moment où l'on connaît mieux, il faudrait accepter de réviser les seuils ». Il n'en a rien été ».
2.2.4.3. Les dioxines
Le pluriel du terme doit être souligné pour un élément dont l'instrumentalisation s'est faite à partir d'un singulier : la dioxine qui produit son effet médiatique, mais n'a pas de signification scientifique. Les dioxines se répartissent en deux catégories qui regroupent plus de 200 molécules différentes. Ils appartiennent à une famille de composés aromatiques polycycliques chlorés présentant des propriétés physico-chimiques semblables. Les toxicologues évaluent à 17 le nombre de dioxines toxiques. La dioxine à laquelle il est fait référence au singulier est celle relâchée lors de l'accident industriel de Seveso 18 ( * ) en juillet 1976 : la 2-3-7-8 TCDD (tétrachlorodibenzo-p-dioxine) classée en 1997 comme « cancérogène certain » par le Centre national de recherche contre le cancer. Les dioxines préexistaient à toute activité humaine ; des sources « naturelles » en relâchant des quantités substantielles telles les éruptions volcaniques et les incendies de forêt. Il semble également que la combustion du charbon en produise beaucoup. Un rapide calcul montre que la totalité des cigarettes fumées en France relâcherait la moitié des dioxines rejetées par les incinérateurs de déchets ménagers. Ces installations se trouvent être au coeur de contestations fondées d'une part sur des peurs entretenues dans des conditions qui n'ont rien de scientifique, en jouant, on l'a vu, sur les termes eux-mêmes et, d'autre part, en instrumentalisant des normes fortement sévérisées au niveau communautaire.
Les incidents qui ont eu lieu sur des incinérateurs hors normes ont donné lieu à des mises en cause systématiques du principe même de l'incinération des déchets ménagers dans le cadre d'une campagne agressive dont les manifestations les plus efficaces ont eu lieu dans d'autres pays européens où le blocage de la construction d'incinérateurs a logiquement entraîné des exportations frauduleuses de déchets dans des pays voisins et même lointains. Or les efforts de mise aux normes dans le cadre des législations française (1991) et européenne actuellement applicables ont apporté une réduction drastique.
Mme Roselyne Bachelot-Narquin alors Ministre de l'écologie et du développement durable, lors de son audition en juin 2003 devant la Délégation à l'aménagement du territoire de l'Assemblée nationale (rapport d'information n° 1169 « Déchets : état d'urgence », 3 novembre 2003) rappelait précisément le caractère satisfaisant des normes actuelles :
« Première priorité : minimiser les impacts des installations de traitement de déchets sur la santé et l'environnement.
C'est sans doute, quand on interroge nos citoyens, la chose à laquelle ils sont les plus sensibles. Il faut en effet qu'étant rassurés sur l'impact de ces installations sur la santé et l'environnement, ils puissent les accepter. Bien gérés et aux normes, ces incinérateurs et ces décharges sont tout à fait acceptables pour la santé et l'environnement.
Dans ce cadre, il s'agit également de lever une confusion, puisque ce sont bien les installations hors normes qui sont visées. Je suis payée pour voir, si je puis dire, puisque j'ai un incinérateur à dioxine dans ma ville, à Angers, qui a fait les titres de la presse nationale pendant les derniers congés d'été : on a parlé en effet de 18 morts par cancer aux alentours de l'incinérateur ! Tout cela ameute évidemment les populations, à juste titre, alors que les installations aux normes, bien entendu, sont sans danger ».
L'InVS avait d'ailleurs été saisi du cas de l'incinérateur d'Angers pour lequel dans son rapport annuel 2002 (page 59), il donne les résultats de son évaluation et remet en perspective l'ensemble du problème :
« Pour les dioxines, les « immissions » modélisées à Angers et imputables à l'usine d'incinération sont comparables avec les teneurs observées en environnement urbain. Avant la mise aux normes, les surexpositions moyennes attribuables à l'incinérateur sont de l'ordre du quart de l'exposition moyenne - à l'époque - de la population française générale. Le ratio de danger est inférieur à 1 ; en considérant l'absence de seuil, l'excès de risque individuel (sur 70 ans) est de 5 sur 10 000 et l'impact sanitaire (sur 25 ans) de 18 cas de cancer. Après mise aux normes, l'excès de risque individuel (sur 70 ans) est de 8 sur 10 millions et l'impact sanitaire (sur 30 ans) inférieur à 1 cas.
De nombreuses incertitudes affectent les résultats concernant le paramètre « dioxines » (peu de mesures d'émission, comportement des substances dans l'environnement, relation dose-réponse). Malgré leur plausibilité et leur cohérence (référentiels environnementaux et autres études menées autour d'incinérateur), ces résultats sont à considérer prudemment.
Les résultats de cette évaluation n'ont pas conduit à préconiser des mesures de prévention particulières. Ils montrent que la mise aux normes de l'incinérateur d'Angers a permis de beaucoup réduire les expositions. Ceci témoigne du bénéfice sanitaire de la mise aux normes des anciens incinérateurs et en particulier ceux qui, comme à Angers, sont situés dans des zones à forte densité de population ».
Malgré les niveaux satisfaisants atteints par la mise aux normes actuelles que cet exemple illustre clairement, le mouvement de « sévérisation » va se poursuivre pour atteindre la valeur fixée par la directive 2000/7G du 4 décembre 2000 : 0,1 nanogramme/m 3 TE toxicity équivalents. L'actuel ministre de l'écologie et du développement durable, M. Serge Lepeltier, l'a ainsi précisé lors du débat organisé à l'Assemblée nationale le 13 avril 2004 sur « la politique de gestion durable des déchets ménagers et assimilés » :
« La gestion des déchets ne peut être durable que si les impacts de leur traitement sur l'environnement et la santé sont maîtrisés. Le cas des incinérateurs d'ordures ménagères est à cet égard instructif. Une réglementation les concernant a été définie au début des années 1990, mais a été mal appliquée - je suis d'accord sur ce point avec M. Sandrier. Sur instruction du ministère en charge de l'environnement, les préfets et les inspecteurs des installations classées se sont ensuite mobilisés pour faire cesser les situations d'infraction. Vous savez la détermination dont a fait preuve le précédent gouvernement, et en particulier Mme Bachelot, pour faire fermer les trente-six unités non-conformes qui restaient encore en fonctionnement il y a deux ans.
Alors que 300 usines, souvent de faible, voire de très faible capacité, fonctionnaient en 1998, moins de 130 sont aujourd'hui en service. Les émissions annuelles de dioxines des usines d'incinération, qui s'élevaient à plus d'un kilo en 1995, sont ainsi retombées à un peu plus de 100 grammes en 2003, soit une division par dix en quelques années. Ces deux chiffres illustrent l'évolution importante et rapide qu'a connue l'incinération en France dernièrement.
Pourtant, il faut encore poursuivre les efforts. Les autres pays européens qui, comme nous, incinèrent une part notable d'ordures ménagères, appliquent en effet depuis plusieurs années des normes de rejets plus strictes, qui seront en vigueur en France fin 2005. Cette nouvelle vague de modernisation entraînera une diminution supplémentaire des rejets de dioxines qui devraient être réduits à 20 gr par an en 2006, soit l'ordre de grandeur que l'on retrouve chez nos partenaires européens.
Les délais sont courts pour faire des travaux importants, mais dans beaucoup de sites, les choses sont bien avancées et je tiens à dire que je serai très ferme vis-à-vis des retardataires. Tout cela aura un coût élevé, j'en suis bien conscient, mais c'est en montrant que l'Etat veille au respect des règles déjà appliquées par nos voisins européens que la confiance dans ce mode de traitement pourra être restaurée ».
Il est à noter que l'argumentation de cette sévérisation, acceptée par la France au niveau communautaire, est essentiellement fondée sur l'alignement sur les pays européens voisins.
Mme Roselyne Bachelot-Narquin avait d'ailleurs eu dans l'audition précitée la même démarche :
« Je veux donc poursuivre cette mise aux normes et, en particulier, respecter l'échéance européenne de 2005 en divisant par dix les quantités de dioxine émises par une action réglementaire déterminée. Je veux surtout accompagner les opérateurs et non pas, comme cela s'est passé de 1992 à 2002, laisser les gens attendre alors qu'il faut les amener à anticiper cette échéance de 2005 ».
Les situations moyennes existantes sous l'empire des règles de 1991 visaient des émissions environ 100 fois supérieures (10 ng/m 3 ) mais les incinérateurs à l'origine de problèmes réels dépassaient eux-mêmes ce seuil, et dans certains cas largement.
Dans le même débat, M. François-Michel Gonnot, Député, montrait par ailleurs que le principe même de l'incinération des déchets était menacé, ce qui montre que la sévérisation des normes a surtout pour but, pour certains, de rendre le plus cher et le plus difficile possible ce mode de traitement :
« Voulons-nous des incinérateurs ? On en sait le coût. Des projets existent. Va-t-on les poursuivre en cherchant à les solvabiliser ? Faut-il classer la valorisation thermique des déchets en énergie propre ? Elle contribue effectivement à diminuer l'effet de serre, et on l'a déjà fait pour le traitement de la biomasse. Alors pourquoi considérerait-on l'incinération des déchets ménagers comme une industrie polluante ? Tel serait pourtant le chemin que l'on prendrait si l'on faisait supporter aux incinérateurs une taxe sur le CO2, comme il en est question ».
Cette menace paraît maintenant écartée, mais le fait qu'une telle orientation ait été envisagée est en soi révélateur. Enfin, pour que l'évolution des traitements puisse se faire sans mouvements chaotiques désorganisant les filières, encore faut-il que ne soient pas autorisées des pratiques qui ont pour but de résoudre des situations particulières tout en prenant des libertés avec les principes d'un respect effectif de l'environnement (enfouissement de déchets ménagers dans les mines de sel comme alternative à l'incinération).
Une dernière observation doit être faite au sujet de cette directive 2000/76. Les remarques précédentes visent la norme dioxines et elle seule, le problème des émissions de métaux lourds dans l'atmosphère étant fort différent et n'ayant pas été abordé dans le même « climat politique ».
2.2.4.4. Le plomb dans l'eau
La toxicité du plomb ne saurait évidemment faire l'objet d'une quelconque controverse. En revanche, la norme dans l'eau potable a été l'objet d'un débat qui, dans l'Union européenne, a abouti à la décision de supprimer le plomb de toutes les canalisations de distribution d'eau.
Il doit être rappelé que si le plomb présente des dangers pour l'homme, c'est essentiellement à travers d'autres modes de contamination que l'eau potable. Il s'agit par exemple des résidus de peinture ingérés par des enfants dans des logements très anciens et insalubres ou, sur une échelle plus large, avec du plomb utilisé comme anti-détonnant dans l'essence. Ce plomb a été supprimé parce que sa présence était radicalement incompatible avec la technologie du pot d'échappement catalytique.
La fixation d'une norme scientifiquement fondée, à un niveau raisonnable, couplée au traitement des eaux qui permet de réduire la solubilité du plomb a été envisagée il y a une vingtaine d'années. On a préféré fixer la norme à 10 g/litre, ce qui entraîne la suppression de toutes les canalisations en plomb.
Le professeur Claude Boudene (toxicologue, membre de l'Académie de médecine), dans une publication de septembre 2001 19 ( * ) , précisait les conditions de rigueur qui devraient présider à cette catégorie de normes dans les termes suivants :
« Un intérêt toxicologique particulier a été apporté, ces dernières années, à la présence de certains contaminants minéraux dans les eaux de boisson, aboutissant à la fixation par l'OMS de valeurs-guides de concentrations qui ont été généralement reprises sous forme de normes par une directive européenne en cours de transposition en droit national.
La démarche intellectuelle qui préside à cette fixation relève de plus en plus de l'application du principe de précaution d'ailleurs désormais reconnu par la Communauté Européenne, dans le but, fort louable, d'assurer un maximum de sécurité au consommateur.
Toutefois, si cette application mérite d'être poussée à l'extrême dans le cas où le dossier toxicologique d'une substance nouvelle est notoirement insuffisant et ne permet pas une évaluation satisfaisante du risque, elle doit être pleinement raisonnée dans le cas d'un toxique ayant déjà fait l'objet de nombreux travaux publiés et reconnus par diverses instances scientifiques. Dans ce cas, la méthodologie utilisée pour cette évaluation doit être discutée de manière approfondie sur la base des données acquises et ne pas céder à des pulsions sentimentales du moment.
L'actualité récente a en effet montré, à propos des deux exemples du plomb et de l'arsenic, combien l'utilisation de modèles mathématiques linéaires, initialement préconisés par l'Agence de Protection de l'Environnement américaine (EPA) et souvent repris par l'OMS dans le cas de molécules potentiellement cancérogènes, risquaient d'aboutir à la fixation de normes dont l'extrême sévérité confinant à l'irréalisme ne vont pas forcément dans le sens d'une amélioration évidente de la santé publique. A un certain stade se pose, en effet, le problème d'une hiérarchisation des risques qui doit obligatoirement tenir compte du coût de l'application de telles mesures dont le bénéfice pour la santé publique n'aura pas été clairement démontré ».
Remettant cette décision européenne en perspective à la fois sanitaire et technologique, l'Office parlementaire d'évaluation des choix scientifiques et technologiques par le rapport de M. Gérard Miquel sur les effets des métaux lourds sur l'environnement et la santé (rapport n°261/2000-2001 Sénat et 2979 Assemblée nationale - 11 ème législation - page 305), apportait les éléments de jugement suivants :
« Le but peut-il être atteint à un coût moindre ? On observera tout d'abord que la suppression des seules canalisations en plomb ne supprime pas le risque hydrique. Il existe des contaminations supérieures à 10 g/l en l'absence de plomb dans les canalisations.
93,5 % de la population n'est guère exposée aux apports de plomb d'origine hydrique. La mortalité liée au plomb est nulle. Seules quelques régions et/ou quelques populations sont à surveiller. « En France métropolitaine, 6000 unités de distribution délivrant à 3,7 millions d'habitants des eaux faiblement minéralisées (pH/6,5), susceptibles d'être en contact avec des canalisations en plomb (Vosges, Massif central ...). En Outremer, ce sont 1,1 million d'habitants pour 212 unités de distribution (Inserm - Le plomb dans l'environnement - 1999).
Une solution consistant à adopter une CMA à 25 g/l associée à une valeur guide objectif à 10 g/l aurait sans nul doute été moins coûteuse, le remplacement des anciennes canalisations s'opérant alors au rythme normal de l'usure, et les sites et populations à risques pouvant bénéficier le cas échéant d'aides ou de programmes spécifiques destinés à alléger la charge en plomb dans l'eau. Une action ciblée paraît toujours plus appropriée qu'une mesure générale.
Enfin, la dépense pouvait-elle être mieux utilisée ? La France va dépenser 70 milliards de francs pour limiter un risque faible. Tandis que dans le même temps il existe des contaminations et des expositions beaucoup plus importantes, beaucoup plus graves (l'exposition liée aux vieilles peintures, l'arsenic dans l'eau ...) qui peuvent être éradiquées pour un coût bien inférieur ».
Le même rapport, en portant l'analyse à un niveau plus général, citait les propos d'un expert recueillis lors de son audition 20 ( * ) :
« Les zones ou produits à surveiller sont en vérité peu nombreux : les batteries, les installations industrielles des deux siècles passés, les peintures dans les habitats insalubres, les décharges sauvages, les zones géologiques acides ... les traitements uniformes sont coûteux et inopérants. Une politique ciblée, modulée serait, de loin, beaucoup plus efficace. Il ne faut pas chercher à réduire les risques partout en dépensant des moyens importants sur des zones où ils n'existent pas ».
Cette observation générale suffira comme conclusion au présent développement ; elle indique d'ailleurs que tout n'est pas réglé et qu'il reste encore beaucoup à faire (« les installations industrielles des deux siècles passés ») pour rétablir ou maintenir un environnement sain, mais par des actions ciblées et non par l'instrumentalisation des normes.
RÉSUME DE LA PREMIERE PARTIE
|
* Les acquis de l'organisation Couronnant une évolution qui avait commencé près d'un siècle plus tôt (en 1902), la loi du 1 er juillet 1998 a établi une architecture adaptée aux exigences qui avaient été identifiées à la suite des crises des années quatre-vingt-dix et plus particulièrement formalisées par les travaux de la mission d'information sénatoriale de 1996-97 ; ces travaux ont abouti à une proposition de loi qui est directement à l'origine des dispositions évaluées aujourd'hui. Les principes d'organisation retenus sont clairs. L'option d'une agence unique du type de la FDA (Food and Drug Administration) américaine ayant été exclue, ce sont des agences propres à chaque domaine qui ont été créées. Le principe de la séparation entre l'évaluation (agences ou instances spécifiques) et la gestion (administrations ministérielles) a été retenue, le cas de l'AFSSAPS constituant une exception délibérée (en effet, en charge des deux fonctions, elle dispose du pouvoir de police sanitaire). L'Institut de Veille Sanitaire est l'instance de base de la sécurité sanitaire et constitue la tête de réseau de toutes les agences et instances spécifiques. Il a une triple mission de surveillance, d'étude et de recommandation. Le CNSS (Comité National de Sécurité Sanitaire) devait être l'élément central de coordination. La DGS (direction Générale de la Santé) recentrée autour de ses fonctions essentielles, exerce la tutelle exclusive ou partagée (AFSSA) des différentes agences. * Les interrogations -- Les principes sur lesquels la sécurité sanitaire est fondée restent toujours globalement pertinents, mais leur portée doit être mieux cernée pour que leur mise en oeuvre soit à la fois réaliste et efficace. Ainsi, le « risque zéro » n'a de sens que déterminé avec précision vis-à-vis d'un danger grave et précis. -- Le principe de précaution n'a de sens et de valeur opérationnelle que s'il est encadré par ses règles d'application : incertitude scientifique réelle, proportionnalité et réversibilité des mesures; en outre l'existence de structures et de mécanismes d'évaluation des risques tels que ceux créés par la loi de 1998 doit avoir pour effet de limiter son champ d'application au fur et à mesure des progrès de la veille et de la sécurité sanitaire. -- Des réalités qui imposent des réorientations Après les progrès considérables qui ont été enregistrés dans le domaine de la sécurité des aliments, on observe la coexistence d'un haut niveau de sécurité sanitaire et d'une atmosphère d'inquiétude croissante ; certaines options semblent en fait calquées non sur la réalité du risque, mais sur les progrès technologiques des appareils de mesure. La sévérisation des normes peut illustrer cette dérive. Les normes relatives aux nitrates ou au plomb dans l'eau potable se sont révélées, par leur niveau, fondées sur des démarches scientifiques erronées pour l'une et arbitraire pour l'autre. Il convient donc d'ajuster les dispositifs d'identification et de mesure des risques à leur gravité réelle, ce qui implique la réévaluation de certains d'entre eux (ultraviolets par exemple). Enfin, le hiatus entre les niveaux élevés de sécurité atteints (domaine alimentaire) et les certitudes de désastres dus à des comportements à risque (obésité) interpellent plus que jamais sur l'orientation et la concentration de la vigilance sanitaire sur les véritables risques. |
DEUXIÈME
PARTIE :
L'AFSSA ET LA SÉCURITÉ SANITAIRE DES
ALIMENTS
En matière de sécurité sanitaire des aliments, la mise en oeuvre de la loi n° 98-535 du 1 er juillet 1998 s'est réalisée principalement avec le décret n° 99-242 du 26 mars 1999. La mise en place de l'AFSSA s'est faite rapidement ensuite, après la nomination de son directeur général. Celui-ci a créé une organisation provisoire de l'Agence en octobre 1999, puis en novembre 2000. Une nouvelle organisation a été pérennisée, les modifications ayant essentiellement touché les structures scientifiques : le nombre de directions opérationnelles a aussi été ramené à trois au lieu de quatre : la DERNS (direction de l'évaluation et des risques sanitaires et nutritionnels, la direction de la programmation des laboratoires et l'ANMV (Agence nationale du médicament vétérinaire).
L'évaluation d'ensemble de l'AFSSA repose sur le résultat global du nouveau dispositif, l'adéquation aux objectifs fixés par une loi très novatrice dans un domaine ou il n'y avait pas d'instance opérationnelle préexistante. Le terme de dispositif doit être souligné car l'AFSSA a pour tâche l'évaluation du risque. Le principe retenu dans le domaine alimentaire a été celui de la séparation entre l'évaluation du risque sanitaire et nutritionnel d'une part, et sa gestion d'autre part, laquelle continue à relever des trois tutelles que sont la DGS, la DGAL et la DGCCRF. L'Agence n'est pas la seule partie prenante dans le domaine de la sécurité alimentaire.
On présentera tout d'abord les éléments de cette évaluation globale du dispositif en rappelant brièvement les objectifs et les principes de la loi de 1998, puis comment l'atteinte de ces objectifs atteste d'un succès incontestable, pour l'Agence en premier lieu.
Dans un second temps, on analysera plus largement comment ce succès appelle néanmoins des confirmations, des ajustements, des clarifications, des ajouts et des mises au point, sans préjudice d'autres retouches.
Enfin, on fera le point sur la dimension européenne du dispositif et de la coordination qu'il implique.
I. UNE ÉVALUATION GLOBALEMENT POSITIVE
1.1. Rappel des objectifs et des principes
? Les crises et les premières réflexions
Les objectifs de la loi de 1998 dans son ensemble découlent directement des crises sanitaires qu'ont connues la France et les pays européens dans le domaine alimentaire : vache folle (ESB), listeria pour les plus importantes. Au-delà de ces événements les plus spectaculaires, des inquiétudes durables avaient attiré l'attention de spécialistes, des responsables politiques et administratifs et des consommateurs eux-mêmes, tel le recours aux antibiotiques dans l'élevage comme facteur de croissance et non comme médicament. Les travaux de la mission d'information sénatoriale sur le renforcement de la sécurité sanitaire en France, présidée par M. Charles Descours et dont M. Claude Huriet était le rapporteur en 1996-1997 et qui sont à l'origine de la loi de 1998, avaient résumé les défauts des dispositions antérieures en des termes précis et dépourvus d'indulgence : « les procédures applicables à l'alimentation ne sont pas satisfaisantes au regard des principes qui doivent guider toute politique de sécurité sanitaire : 1/la réglementation applicable aux produits alimentaires n'est pas nécessairement orientée vers la protection de la santé de l'homme 2/le principe de précaution n'est pas toujours appliqué, l'indépendance des contrôles est insuffisante ».
Les termes même du rapport de cette mission indiquent comment dès l'origine la nouvelle agence a été conçue (Sénat n° 196 (1996-97 page 65) :
« Votre commission propose la création d'un nouvel établissement public, l'Agence de sécurité sanitaire des produits alimentaires. Elle aurait une double mission : d'une part, assurer le contrôle et garantir la sécurité des produits alimentaires, au lieu et place des services du ministère de l'agriculture et de l'alimentation et du ministère de l'économie et des finances et, d'autre part, assurer le contrôle du médicament vétérinaire, à la place de l'Agence du médicament vétérinaire qui y serait intégrée.
Cette Agence serait placée sous la tutelle des ministères de la santé, de l'agriculture, de l'économie et des finances et de l'industrie.
Elle aurait compétence pour proposer aux ministres chargés de la santé et de l'agriculture toute modification de la réglementation concernant la sécurité sanitaire des produits alimentaires.
Il serait ainsi mis fin à ce qui peut s'analyser comme un « mélange des genres » et qui est préjudiciable, tant à la confiance des consommateurs qu'à la réalité de la sécurité sanitaire des produits alimentaires.
Au sein de l'Etat, la légitime représentation des intérêts des producteurs serait ainsi séparée de celle des intérêts de la santé publique, non moins légitimes mais parfois contradictoires des premiers ».
? Les acquis antérieurs
Il ne faudrait cependant pas laisser croire que l'on partait de rien et que l'Etat s'était complètement désintéressé de ces questions. Par rapport à d'autres pays européens, on peut même considérer que le niveau de sécurité en France se comparait favorablement. Par exemple, le nombre d'intoxications aux salmonelloses y était plutôt bas et, par ailleurs, on n'y avait pas connu des crises d'une gravité telle que celle de l'huile d'olive frelatée (Espagne) ou du vin blanc édulcoré au glycol (Autriche) sans parler plus tardivement des volailles contaminées par des dioxines (Belgique).
Le Comité Supérieur d'Hygiène Publique de France, dont la création remonte à une loi de 1902, à travers de l'une de ses quatre sections spécialisées « alimentation et nutrition » était, avant la loi de 1998, en charge des expertises maintenant réalisées par l'AFSSA. Il avait contribué à atteindre un niveau qui n'était certes plus satisfaisant au regard des exigences et des défis des années quatre vingt dix, mais qui avait permis d'éviter le pire. Un nombre substantiel des experts qui le constituaient a d'ailleurs été retenu après une sélection sévère lors de la constitution des premiers comités d'experts de l'AFSSA en 2001.
Par ailleurs, les trois administrations en charge des contrôles ont contribué substantiellement, à maintenir une pression qui s'est souvent révélée fort efficace pour éviter ou limiter certaines crises. Mais à l'évidence le cadre administratif n'était plus adapté et la culture de l'évaluation du risque ne pouvait pas vraiment s'y développer.
Pour assurer un niveau élevé de sécurité sanitaire des aliments en rapport avec les risques constatés et les défis identifiés, plusieurs nécessités ont été particulièrement pointées : la rationalisation, la réactivité et l'indépendance. Il s'agit des objectifs principaux qui sous-tendent ensuite une organisation et des moyens à leur hauteur. C'est dans ces perspectives que l'AFSSA a été conçue au niveau parlementaire et gouvernemental, puis construite par les responsables gouvernementaux et administratifs au premier rang desquels sa direction. C'est aussi dans cette perspective qu'elle est gérée et s'est développée. La concertation prévue par la loi avec les autres parties prenantes dans ce domaine fait aussi partie de son cadre d'action.
1.2. Les choix du législateur
1.2.1. Une agence spécialisée
Le premier point de discussion apparu dès le début des travaux de la mission d'information de la commission des affaires sociales du Sénat en 1996 sur l'ensemble de la sécurité sanitaire a été celui du choix : agence unique ou agences spécialisées par risque ou par domaine. En première approche, la FDA américaine (Food and Drug Administration) sans s'imposer vraiment comme modèle, apparaissait comme une référence incontournable à partir de laquelle les différents schémas pouvaient s'ordonnancer. Malgré l'intérêt évident de cette instance et de ses modes de travail, il s'est rapidement avéré qu'il y avait beaucoup d'idées inexactes, sinon fausses, sur la FDA. En outre, compte tenu des différences considérables de tous ordres entre les Etats-Unis et la France, elle ne pouvait pas être considérée comme un modèle. Or sur la question de l'agence unique détenant l'ensemble des compétences, plusieurs mises au point doivent préalablement être faites.
Ainsi la FDA est en charge des produits destinés à l'homme : produits thérapeutiques et produits alimentaires principalement. Mais, dans ce dernier domaine, le contrôle de la viande et des oeufs est assuré par le département de l'agriculture ; en outre, l'Agence fédérale de l'environnement intervient de son côté sur certains points.
Plusieurs interlocuteurs responsables ou anciens responsables américains avaient à l'époque eux-mêmes estimé que « si c'était à refaire, on créerait deux agences au lieu d'une seule ».
Le principe de l'agence unique pour les produits de santé et les aliments a des défenseurs même après l'affaiblissement de « l'argument américain ». Mais au fur et à mesure de l'élaboration de la réflexion, la proposition a été abandonnée. Le changement de majorité à l'Assemblée nationale suite à la dissolution en avril 1997 n'entraînera pas de longue interruption comme on aurait pu le craindre, mais la perspective de l'agence unique redevint d'actualité. M. Claude Huriet, lors de la journée d'audition du 29 mai 2000 à la commission des affaires sociales du Sénat rappela dans les termes suivants cette nouvelle phase de réflexion qui aboutit à la situation que nous connaissons avec deux agences :
« Après le changement de gouvernement, le nouveau Premier ministre, M. Lionel Jospin, a évoqué la création d'une Agence de Sécurité Sanitaire dans sa déclaration de politique générale au Parlement. D'ailleurs, pendant que nous présentions notre proposition de loi sous le gouvernement précédent, notre ami Bernard Kouchner avait écrit un article dans lequel il défendait la création d'une seule agence, alors que nous en proposions deux.
Nous avons écouté ce que disait M. Jospin et, le 12 août, des conseillers de M. Jospin ont demandé à nous rencontrer pour parler de cette affaire. Cela montre qu'au-delà des affrontements normaux en démocratie, de grandes questions transcendent les courants politiques.
Le cabinet du Premier ministre nous a indiqué que notre proposition de loi figurerait à l'ordre du jour du Parlement.
Ils nous ont cependant suggéré de créer une agence plutôt que deux. Nous leur avons dit : « Vous allez voir que faire une agence est plus compliqué que ce que vous croyez. Si vous arrivez à faire une agence, nous n'en faisons pas une affaire de paternité, nous voulons bien n'en faire qu'une, mais vous verrez que vous serez obligés d'en faire deux ».
C'est là une question sur laquelle il n'y aura guère à revenir en détail car, a priori, aucun argument ne conduit aujourd'hui à revenir sur ce choix qui s'est révélé être le bon. D'ailleurs, certaines évolutions ou réformes internes dans d'autres pays vont dans le même sens.
1.2.2. La séparation entre l'évaluation et la gestion du risque.
Thème important, spécifique au domaine alimentaire, ce principe est préconisé par le Codex alimentarius, organisme placé sous l'égide de la FAO et de l'OMS. Mais sa nécessité a surtout été soulignée à partir de certains contre-exemples mettant en cause des administrations ou des instances d'expertises ou autres dans lesquelles on avait relevé un regrettable mélange des genres : représentation de branches économiques et évaluation du risque.
Il avait été envisagé que l'AFSSA se voit doter, au-delà de l'évaluation, de la gestion des risques, comme c'est le cas pour l'AFSSAPS, mais le principe de la séparation a prévalu. Ce thème qui est revenu parfois en discussion, sera analysé en deuxième partie de cette évaluation.
1.2.3. Le format de l'Agence
Une conception défendue avant 1998 aurait fait de l'Agence une entité légère, dont le principal rôle aurait été de structurer les diverses instances d'expertises existantes alors, mais qui étaient très dispersées et fonctionnaient souvent dans des conditions rudimentaires, notamment en raison de l'évidence insuffisance de moyens.
C'est une conception beaucoup plus ample qui a prévalu, notamment avec l'intégration complète des laboratoires du CNEVA (centre national d'études vétérinaires et alimentaires) et de l'ANMV (Agence nationale du médicament vétérinaire). Cette intégration, qui n'était pas prévue à l'origine, a de l'avis général, donné la masse critique à l'AFSSA lui permettant de répondre aux demandes qui lui sont adressées et aux objectifs qui lui sont fixés.
Il doit être rappelé que dans le domaine du médicament vétérinaire, les mécanismes n'ont pas été modifiés par rapport à la situation antérieure et que, par conséquent, l'AFSSA dispose d'un pouvoir de police sanitaire et est donc compétente pour la gestion du risque et la fonction de contrôle. Cette exception au principe en vigueur dans le domaine des produits alimentaires eux-mêmes s'explique aisément au niveau des principes et à celui de la mise en oeuvre sur le terrain : on est d'ailleurs là dans une situation très comparable à celle de l'AFSSAPS avec le médicament humain.
1.2.4. Les domaines affectés à l'AFSSA
Le cas du secteur vétérinaire venant d'être précisé, le domaine général de l'Agence est donc large : il concerne tous les produits alimentaires « de la fourche à la fourchette ». L'agence dispose en outre des moyens nécessaires à sa mission d'évaluation par les laboratoires et ses services techniques et à travers les 10 comités d'experts qu'elle anime.
1.2.4.1. Les missions de l'AFSSA
Précisées pour l'essentiel par le code de la santé publique en ses articles L. 1323-1 et L. 1323-2, ainsi que le R. 794-2, elles sont principalement au nombre de trois, cette classification n'étant pas indiscutable :
-- la mission d'évaluation des risques sanitaires et nutritionnels.
Couvrant donc l'ensemble de la chaîne alimentaire, cette mission :
concerne chacune des étapes de cette chaîne : production, transformation, conservation, transport, stockage et distribution ;
s'applique aussi bien aux aliments destinés à l'homme qu'aux aliments destinés aux animaux, qu'ils soient d'origine animale ou végétale, mais aussi les eaux destinées à la consommation humaine ;
outre les aliments eux-mêmes, l'évaluation englobe les différents produits dont l'utilisation peut avoir des conséquences sur la sécurité des aliments : produits phytosanitaires, médicaments vétérinaires, matières fertilisantes, matériaux en contact avec les aliments ou produits de conditionnement ;
Toutefois, l'existence de « zones frontières » rendent plus floues les compétences de l'Agence en ce qui concerne notamment les « intrants » : produits phytosanitaires, engrais et les OGM où il n'y a pas là exclusivité de l'évaluation. D'importantes difficultés ont pour origine ces configurations incertaines et justifient des propositions adaptées.
Il convient également de souligner que cette mission vise les risques nutritionnels, au-delà des risques sanitaires.
-- la mission d'appui scientifique et technique
L'appui scientifique et technique comporte principalement la recherche et l'expertise pour l'élaboration et l'application de la réglementation sanitaire, de méthodes diagnostiques et thérapeutiques et la mise au point d'essais et de contrôles.
Elle recouvre les missions qui incombaient auparavant au CNEVA. Elle s'organise autour des 13 laboratoires qui lui étaient rattachés (les 12 du CNEVA et le laboratoire d'hydrologie et d'études thermales). Ces missions étaient définies par le décret du 29 avril 1988, portant création du CNEVA et comportaient :
le soutien scientifique et technique à l'élaboration, à l'application et à l'évaluation des mesures prises par le ministre de l'agriculture, ou d'autres ministres intéressés, dans les domaines du médicament vétérinaire, de la santé animale, du bien-être des animaux et de leurs conséquences sur l'hygiène publique, ainsi que de la sécurité de l'alimentation humaine liée à la consommation de denrées d'origine animale ;
la conception et l'utilisation de nouvelles techniques ;
l'établissement de normes applicables aux aliments pour valoriser leur qualité ;
la mise en oeuvre et le développement de programmes tendant à la protection sanitaire et à la salubrité alimentaire.
Les activités menées dans les laboratoires de l'AFSSA dans le cadre de l'appui scientifique et technique ne coïncident donc pas avec les domaines de compétence de l'agence en matière d'évaluation des risques, mais elles sont très largement complémentaires.
-- la recherche
Si la recherche n'est pas nécessairement une mission de l'Agence en soi, elle est constitutive de plusieurs de ses activités et prioritairement, bien sûr, de l'appui scientifique et technique qui lui est indispensable. Elle ne saurait toutefois se limiter à cela. C'est toute l'ambiguïté de la place et de l'orientation de la recherche à l'AFSSA et son l'articulation avec la recherche menée par d'autres entités administratives dans ces domaines alimentaires et nutritionnels. Une amélioration des synergies doit donc être envisagée.
1.2.4.2. L'évaluation de la gestion du risque
L'évaluation de la gestion du risque est apparue comme une mission propre que caractérisent l'évaluation des études et contrôles réalisés par les services ministériels (art. L. 1322-2 9°), le soutien et la concertation mutuelles que l'AFSSA et les trois tutelles ont à assurer notamment à travers les plans de surveillance et de contrôle, la communication des résultats des contrôles et enquêtes. Cette mission n'est plus aujourd'hui identifiée d'une manière autonome, ses éléments constitutifs sont souvent présentés comme le prolongement de l'évaluation du risque (pour les plans de surveillance et de contrôle) et comme une part des échanges au titre de l'appui scientifique et technique. Il s'agit là d'un ensemble de modalités d'action où des difficultés demeurent et où des mises au point sont à réaliser.
1.3. La réalisation des objectifs : un succès incontestable
1.3.1. L'appréciation globale
Par rapport aux objectifs plutôt ambitieux que la loi de 1998 a fixés, l'AFSSA a, de l'avis général, atteint l'essentiel de ceux qui lui ont été fixés. Cette constatation est le fait tant des connaisseurs les plus expérimentées du domaine, experts, hauts fonctionnaires, responsables et anciens responsables que de ceux, extérieurs aux circuits administratifs, qui ont a en connaître par leurs fonctions professionnelles ou associatives : monde agricole, industrie, distribution, associations de consommateurs ; en outre, la considération générale dont l'AFSSA jouit s'observe également chez des acteurs et observateurs plus lointains au niveau européen dans les pays voisins.
Les acquis préexistants à la loi de 1998 ont été réunis avec profit. La nouvelle structure, la seule qui dans cette réforme d'ensemble a été être créée ex-nihilo, a rapidement pris corps en atteignant d'emblée un niveau opérationnel. La dotation en moyens, humains notamment, était l'une des conditions de cette efficacité ; elle a été convenable ainsi que les travaux des inspections l'ont vérifié. Par exemple, l'augmentation significative des effectifs consacrés à l'organisation d'une expertise a été de pair avec le recrutement de personnel scientifique de niveau élevé et spécialisé.
La crédibilité de l'Agence dans son ensemble s'est rapidement établie notamment en tant qu'autorité indépendante de l'évaluation des risques. Dans l'ensemble du domaine de compétences fort large qui est le sien, ses avis sont attendus, appréciés et rendus dans des conditions réellement satisfaisantes, de rapidité notamment. La constitution et l'animation des comités d'experts spécialisés recueillent aussi des appréciations favorables et les comparaisons que l'on peut faire sous cet éclairage sont à l'avantage de l'AFSSA.
L'usage du droit d'auto-saisine a été apprécié par de nombreuses personnes consultées. Les échanges et les productions auxquels il a donné lieu ne sont néanmoins pas exempts de critiques qui mettent en cause la concrétisation d'un droit dont le principe reste incontestable ; la communication qui l'accompagne fait l'objet de quelques reproches parfois justifiés.
La communication de l'Agence est d'ailleurs l'un des thèmes qui soulève le plus de controverses. On en donnera quelques indices. Mais il est clair que la volonté d'établir une nouvelle autorité autonome, et même indépendante, dans l'évaluation des risques, située sous la tutelle de trois directions générales ministérielles plaçait d'emblée les responsables, et principalement le directeur général de l'AFSSA, dans une situation délicate. La personnalisation dont il fait preuve correspond ainsi à une nécessité et comporte de fait des risques. On ne saurait donc s'étonner que dans le cadre général d'un succès très tangible, le risque se soit concrétisé à travers des positionnements, des annonces et des chronologies parfois discutables, en tout cas sujets à interrogations.
L'autonomie de l'Agence, souhaitée par tous à l'origine et encore aujourd'hui, a un prix. Cela posé, le risque de la communication doit être le plus limité possible et certaines leçons que l'on pourra tirer de l'expérience de cinq années ne doivent pas être négligées.
1.3.2. L'atteinte des buts principaux
* La DERNS : son fonctionnement
La DERNS, direction de l'évaluation des risques nutritionnels et sanitaires, est au coeur de l'activité de l'Agence : c'est la structure nouvelle, spécifique, qui a été créée avec l'Agence. Son rôle, son organisation et ses moyens doivent être précisés pour pouvoir mieux saisir l'ensemble du mécanisme. Elle assure les actions d'évaluation dans le domaine des risques nutritionnels et sanitaires en faisant appel aux comités d'experts spécialisés et aux groupes de travail constitués auprès de l'Agence, aux compétences scientifiques dont elle dispose parmi ses personnels, en travaillant en liaison avec les autres directions de l'Agence.
A ce titre, la DERNS est notamment chargée d'instruire, dans le champ des risques nutritionnels et sanitaires, l'ensemble des demandes d'avis ou de consultations adressées à l'Agence. Elle assure le recueil des informations nécessaires à ses missions et élabore, dans son domaine de compétence, les recommandations ou propositions aux autorités compétentes.
Elle est chargée du secrétariat des comités d'experts spécialisés et des groupes de travail, à l'exception de ceux dont le secrétariat est assuré par l'Agence nationale du médicament vétérinaire ou par le directeur de la santé animale et du bien-être des animaux. Elle est composée des unités suivantes :
- Unité d'appui scientifique et technique à l'expertise
- Unité d'appui épidémiologique à l'analyse du risque
- Unité chargée de l'évaluation sur la nutrition et les risques nutritionnels
- Unité chargée de l'évaluation des risques biologiques
- Unité chargée de l'évaluation des risques physico-chimiques
- Unité chargée de l'évaluation des risques liés à l'eau
- Observatoire des consommations alimentaires et Centre informatique sur la qualité des aliments.
* La DERNS : les experts
La DERNS compte soixante-dix personnes, en majorité des scientifiques, ce qui lui permet de procéder à une expertise interne qui vient en support de l'expertise réalisée ensuite par les comités d'experts spécialisés. Dans certains cas, l'expertise est réalisée seulement en interne, mais il est clair que cela ne doit avoir lieu qu'à titre exceptionnel et lorsque l'objet de l'expertise le permet.
Les activités d'appui du travail des experts sont essentielles et, de l'avis général, bien menées.
L'animation et la procédure dans les comités d'experts spécialisés sont également assurées dans des conditions satisfaisantes et dans lesquelles le secrétariat scientifique, par un membre de la DERNS, joue un rôle essentiel. Mais les critiques ne sont pas absentes ici. Celles qui visent les groupes de travail, notamment dans le cadre d'auto-saisines sur des sujets de fond, de par leur spécificité, seront abordées après le présent développement. En revanche, les critiques sur la lenteur ou l'opacité du calendrier de travail liées au processus d'expertise proprement dit ne paraissent pas dénuées de fondement ; elles sont exprimées nettement par les professionnels.
Les experts externes, bien qu'appréciant l'appui et l'animation des comités spécialisés formulent quelquefois des critiques qui peuvent paraître contradictoires, mais ne le sont pas nécessairement. Ainsi, on a noté que dans un comité, le secrétariat scientifique a du mal à rester dans son rôle et tente de modifier le projet d'avis des experts. Il a été signalé dans un autre domaine que l'aide apportée aux experts par le secrétariat scientifique était insuffisante et que de ce fait la charge de ceux-là devenait de plus en plus lourde en raison de la nécessité d'avoir une rédaction des avis de plus en plus serrée pour éviter toute interprétation douteuse.
Enfin, mais cela n'est pas élément spécifique à l'AFSSA, les conditions et l'atmosphère de travail varient beaucoup d'un comité à l'autre et les « effets rapporteur ou président » sont signalés par de nombreux participants, risquant parfois de limiter excessivement les échanges et les délibérations.
Les conditions d'examen dans les comités pourraient donc être améliorées concrètement : cela fera l'objet de quelques remarques et propositions dans le deuxième développement de la présente partie. Mais on peut déjà indiquer que des modes d'organisation plus efficaces pour une plus grande densité des travaux pourraient s'inspirer des comités d'experts du JECFA (Joint expert committee on food addictives), l'un des trois comités d'experts de la commission du Codex alimentarius (FAO-OMS). Il est à noter d'ailleurs que dans le domaine alimentaire, la France fournit aujourd'hui au niveau international proportionnellement moins d'experts que le Danemark, la Grande-Bretagne ou les Pays-Bas ; les capacités d'expertise des instances de ces pays, dont certaines ont fait l'objet d'ajustement récents, sont en effet appréciées. Un renforcement de la présence des experts français dans les instances internationales semble indispensable.
Des avis qui font référence
Les avis d'évaluation du risque rendu par l'AFSSA, « production centrale » de l'Agence, constituent des références appréciées que l'ensemble des observateurs s'accorde à considérer comme un progrès tangible. Si les objectifs généraux sont atteints, il reste que certains avis, rares mais voyants, ont donné lieu à des controverses qui n'étaient pas injustifiées. Selon les cas, il s'agissait des modalités de communication de l'avis, de sa rédaction, avec notamment le problème de la marge laissée au gestionnaire du risque, décideur ministériel, dans l'espace intellectuel et dans le temps médiatique. L'essentiel de ces questions sera abordé ultérieurement. Toutefois, pour que le signalement ne soit pas incomplet, on se doit de rappeler l'existence de quelques avis controversés.
Il ne saurait être question, bien sûr, d'apprécier ici la valeur et le sens de l'expertise, base de l'avis, mais les échanges qui ont eu lieu ou les différentes positions sont à rappeler. Un exemple « incontournable » doit être présenté ici. L'avis du 25 juin 2001 sur « l'évolution possible des modalités d'abattage des troupeaux » (de bovins dans lesquels un cas d'ESB a été diagnostiqué) est incontestablement celui qui a entraîné les controverses les plus vives et où l'autorité de l'AFSSA a été le plus visiblement contestée.
La Cour des Comptes, dans son relevé de conclusions définitives sur la mise en place de l'AFSSA (en 2002) en donnait la présentation ci-après :
« Un exemple particulièrement significatif est représenté par l'avis émis le 25 juin 2001 relatif à l'abattage total ou partiel des troupeaux de bovins dans lesquels un cas de vache folle avait été diagnostiqué.
L'agence a rendu un avis, qui est apparu particulièrement confus aux tutelles, tout en outrepassant le simple domaine de l'évaluation du risque. Face à cette situation, le ministre de l'agriculture a décidé de saisir le Conseil National de l'Alimentation (CNA) 21 ( * ) et, sur la base de l'avis émis par celui-ci, a considéré qu'il convenait de s'orienter vers l'abattage sélectif mais de préparer les conditions de sa mise en oeuvre et donc de maintenir temporairement, pendant un délai de l'ordre de six mois, la décision d'abattage total de ces troupeaux, tout en indiquant qu'un nouvel avis serait demandé à l'AFSSA quelques mois plus tard sur la base d'un projet de texte.
L'agence a, en effet, été saisie par le ministère de l'agriculture et de la pêche, le 19 décembre 2001, sur un projet d'arrêté.
Elle a, cette fois, émis un avis favorable à ce que le ministre signe un arrêté mettant fin à l'abattage systématique des troupeaux dans lesquels un cas d'ESB a été détecté, notamment en excluant de l'abattage les animaux nés après le 1 er janvier 2002 ; l'AFSSA s'appuyant sur le fait que, dans la saisine, l'administration considérait que, compte tenu des différentes mesures réglementaires prises, elle pouvait exclure tout risque de contamination par voie alimentaire pour les animaux nés à compter de cette date.
Le ministre, avant de prendre sa décision, a décidé de consulter de nouveau le CNA. Celui-ci, tout en approuvant le principe de l'arrêté a, pour tenir compte des conditions réelles d'élevage et de commercialisation des animaux, soulevé des points qui n'avaient pas été prévus par l'arrêté. Il a été suivi par le ministre ».
De son côté, M. Martin Hirsch (directeur général de l'AFSSA), a fait part en 2002 de ses impressions sur les questions les plus marquantes qu'il avait déjà eu à connaître depuis 1999 dans un livre « Ces peurs qui nous gouvernent - Sécurité Sanitaire, faut-il craindre la transparence ? » 22 ( * ) . Consacrant un chapitre d'une dizaine de pages à cet avis, il le présente tout d'abord ainsi :
« Le 25 juin 2001, l'Agence française de sécurité sanitaire des aliments a transmis au gouvernement et rendu public un avis sur « l'évolution possible des modalités d'abattage des troupeaux ». Cet avis indiquait qu'il était possible de suivre une démarche par étapes permettant de faire évoluer les stratégies d'abattage tout en maintenant le même niveau de sécurité pour le consommateur.
Cet avis n'avait rien d'inquiétant. Il ne recommandait pas une mesure supplémentaire à mettre en oeuvre. Il ne mettait pas l'accent sur un risque que l'on n'aurait pas vu jusqu'à présent. Eh bien, pourtant, que de colères provoquées par cet avis ! Il a été vilipendé. Le ministre de l'Agriculture l'a lui-même qualifié de « pas clair » et d'avis dont on ne pouvait rien tirer. Le nouveau président de la FNSEA de « très confus ». Les responsables de la Confédération paysanne de « décevant » ... La question était toute simple : quand on trouve un cas d'ESB dans un troupeau, faut-il abattre la totalité du troupeau ? ou peut-on n'en abattre qu'une partie ? L'abattage total avait été décidé en France lorsque étaient apparus les premiers cas. Il présentait alors plusieurs avantages. Compte tenu des doutes sur l'origine de la maladie, il permettait de ne pas négliger un phénomène particulier qui se serait passé dans un élevage et aurait exposé plusieurs animaux à la maladie. Il montrait la détermination dans la lutte contre l'ESB, par son côté radical, aisément compréhensible. Il donnait l'espoir d'accélérer l'éradication de la maladie dans le cheptel français. La situation est devenue plus compliquée quand l'augmentation du nombre de cas a impliqué un nombre élevé d'abattages, quand les éleveurs ont indiqué que l'abattage total avait quelque chose d'infamant et de pénalisant, et quand les montants annuels des indemnités ont commencé à peser sur les finances publiques. (...)
Cet avis présentait plusieurs particularités, par rapport aux six cents avis que l'agence avait rendus depuis sa création en avril 1999 et à la quarantaine d'avis rendus sur l'ESB. Si ces derniers ne sont pas les plus nombreux, ils ne sont pas les plus simples ni à l'élaborer, ni à interpréter, ni parfois à suivre. Et qu'il s'agisse de l'avis défavorable rendu sur la levée de l'embargo britannique à l'automne 1999, de l'avis recommandant d'étendre la liste des produits bovins (dont les boyaux) au printemps 2000, des premiers résultats des tests sur les animaux à risque à l'automne 2000 ou de l'avis sur les mesures de précaution chez les moutons, les exemples ne manquent pas de difficultés soulevées par le choix de la transparence. Publication obligatoire, d'ailleurs, par la loi. Ces difficultés étaient souvent liées au sentiment que face à un problème pour lequel des mesures importantes avaient déjà été prises, sans que le recul permette d'en apprécier l'efficacité, le fait de les renforcer était davantage perçu comme de l'acharnement de précaution que comme la réponse à une nécessité de prévention. Et que, décidément, le prix à payer pour cette maladie, à tous les sens du terme, était bien élevé.
L'avis sur l'abattage a soulevé, lui, de tout autres questions. Cette fois - pour une fois, auraient pu dire certains ! - l'Agence ne recommandait pas une mesure supplémentaire, mais envisageait un assouplissement dans l'une des mesures actuellement en place. Et pas n'importe laquelle : la première mise en oeuvre lors de l'apparition du premier cas en France ; la mesure la plus symbolique ; la mesure la plus facile à comprendre. Abattre la totalité d'un troupeau, cela frappe plus les esprits que retirer les ganglions para vertébraux chez les animaux pour lesquels un test est négatif et qui sont censés ne pas avoir été nourris avec des farines animales ».
Après ces éléments d'explication du problème et du positionnement du débat, M. Martin Hirsch résume d'autres données plus détaillées, à la lumière des évolutions en cours à l'époque, puis il justifie le cadrage de l'avis rendu en juin 2001 dans ces termes :
« C'est dans ce contexte que les scientifiques ont estimé que l'on pouvait tendre vers un abattage sélectif par des mesures « graduées ». Et ils ont décrit les deux bornes possibles de ce que pouvait être un tel abattage : d'un côté, un abattage « subtotal », seuls les veaux nés après la sécurisation effective de toute l'alimentation animale étant exclus ; de l'autre, un abattage du seul animal malade.
Sur cette base, l'Agence n'a pas formulé de recommandations, contrairement à ce qu'elle fait régulièrement, en application de la loi, quand elle estime que l'analyse d'un risque rend nécessaire de proposer une mesure supplémentaire. Mais, sachant que si le gouvernement décidait de faire évoluer l'abattage total, l'Agence serait obligatoirement amenée à se prononcer sur le nouveau projet de réglementation, elle a indiqué à l'avance dans quelles conditions elle le ferait en cherchant à montrer les différentes options qui, pour les scientifiques, permettaient de maintenir un niveau équivalent de sécurité pour le consommateur ».
Ce cas d'école sur un problème très complexe mais très médiatisé, illustre le danger de solliciter quelque peu la notion « d'évaluation du risque » pour l'Agence même si, se plaçant dans la perspective d'une situation d'évidence évolutive, elle souhaitait en quelque sorte borner pour l'avenir l'évaluation du risque dans un nouveau dispositif.
Très logiquement, à la suite de cette controverse, la Cour des comptes soulignait « l'intérêt d'une procédure claire, ne laissant pas la place aux interventions discrétionnaires de la part des diverses autorités compétentes dont chacune doit intervenir dans son domaine propre, afin d'éviter qu'une instance puisse apparaître comme la chambre d'appel de l'autre ».
Les avis de l'Agence dans d'autres domaines ont pu être mis en cause par exemple sur la méthodologie scientifique (complément de créatine en 2000) ou par la portée réelle de l'avis avec celui « relatif à la réactualisation de la liste des matériaux à risque spécifié chez les ovins et les caprins » du 14 février 2001. L'AFSSA recommandait d'écarter de la consommation humaine les intestins de tous les animaux, quel que soit leur âge et le statut sanitaire du troupeau dont ils proviennent.
Ce dernier cas fut à l'origine d'une violente polémique puisque le Président de la République, inaugurant le Salon de l'Agriculture trois jours plus tard, reproche à l'AFSSA d'avoir crée la panique sur le mouton. Les propos du Président sont sans appel : « les responsables de l'AFSSA sont des irresponsables. Ils ont fait preuve au moins de bêtise et de mauvais goût. Que les scientifiques parlent quand ils ont quelque chose à dire » 23 ( * ) .
Concrètement, il s'agissait pour l'essentiel des intestins d'ovins qui ne sont pas consommés en l'état, mais après un traitement mécanique sont utilisés en tant que boyau pour la production de saucisses ou de merguez. Cette recommandation procédait clairement du principe de précaution, le risque n'étant pas avéré, mais son existence n'étant pas non plus exclue. Dans un avis du 8 novembre 2001, l'AFSSA maintint ses recommandations de l'avis de février 2001 suggérant, en outre, si les intestins ne sont pas retirés de la consommation humaine, qu'une identification soit prévue pour l'information du consommateur. Le comité d'experts spécialisés sur les ESST continuait d'ailleurs ses travaux sur ce sujet.
Ces avis de l'AFSSA s'inscrivent dans la longue série qu'elle est amenée à rendre sur saisine des administrations de tutelle, notamment sur les ESST. Ciblant précisément la réponse, il n'appelait pas une réaction brutale, y compris dans le contexte si particulier du Salon de l'Agriculture et sous la pression des lobbys économiques sur les responsables politiques. Cet événement illustre la difficulté de l'exercice de définition et de communication des avis dès lors qu'une médiatisation aléatoire fait tomber le projecteur sur une précision factuelle qui ne mérite pas cet excès d'honneur.
Ces avis sur les « intestins d'ovins » posaient néanmoins un problème quant à leur portée réelle compte tenu de la situation concrète du secteur : les abattages nationaux ne représentent que 40 % dans les viandes ovines consommées en France. C'est précisément l'objection que fit le Conseil National de l'Alimentation (CNA) le 10 janvier 2002 dans son « avis sur les conséquences socio-économiques du renforcement des mesures de précaution face au risque de ESST chez le mouton » :
« Outre les difficultés juridiques éventuelles à prendre des dispositions nationales isolées, les membres du Conseil, compte tenu de la très forte internationalisation des courants commerciaux pour cette filière, des lacunes existant dans l'identification et la traçabilité des animaux, et de la difficulté particulière à contrôler des produits transformés, s'interrogent sur l'efficacité de mesures nationales qui ne seraient pas relayées par des dispositions identiques aux plans communautaire et international. Leurs interrogations valent à la fois pour les mesures de police sanitaire et de santé publique. Ils se demandent, compte tenu du niveau des importations de produits ovins et des possibilités d'augmentation de ces importations, si une mise en oeuvre unilatérale des préconisations de l'AFSSA conduirait à une réduction effective du risque pour les consommateurs ».
Par ailleurs, « ils s'interrogent sur la contribution de cette mesure de précaution, prise isolément, à la réduction effective des risques qui seraient liés à la présence d'ESB dans le cheptel ovin si elle venait à être démontrée. Si cette limitation de l'avis résulte des circonstances mêmes dans lesquelles il a été rendu, le Conseil estime nécessaire de le signaler (...) ».
« Enfin, certains membres s'interrogent sur la cohérence entre la mise en oeuvre de mesures de précaution au regard du risque d'ESST et le rétablissement parallèle de l'autorisation d'importation de viandes ovines britanniques motivé par la fin de l'épizootie de fièvre aphteuse, alors même que la prévalence de l'ESB en Grande-Bretagne demeure très forte ».
La mesure recommandée par l'AFSSA pouvait donc se révéler excessivement limitée dans ses effets.
1.3.3. Le positionnement nutritionnel de l'AFSSA
* La compétence de l'AFSSA
La compétence générale de l'AFSSA sous l'angle nutritionnel indiqué à l'article L 1323-1 du code de santé publique est précisée sans ambages à l'art. L 1323-2-6 : « l'Agence évalue la pertinence des données spécifiques transmises en vue de fournir une expertise sur les propriétés nutritionnelles et fonctionnelles des aliments ... ». L'AFSSA accorde depuis l'origine toute l'importance qu'elle mérite à l'accomplissement de ces tâches dans ce domaine vaste et essentiel. L'Agence précise ainsi elle-même la prise en charge et le traitement de ces questions (rapport d'activité 2001-2002) :
« L'unité d'évaluation sur la nutrition et les risques nutritionnels est chargée d'évaluer les risques nutritionnels et les propriétés nutritionnelles et fonctionnelles des substances et denrées entrant dans l'alimentation humaine, de proposer des avis et recommandations sur des grands thèmes de santé publique dans le domaine de la nutrition, d'élaborer des références nutritionnelles dans le cadre d'une politique de santé publique, et de participer à la mission d'information de l'Agence.
L'unité nutrition assure le secrétariat scientifique du comité d'experts spécialisé « Nutrition » dont les compétences couvrent les principaux aspects de la nutrition humaine, dans ses dimensions métaboliques ou psycho comportementales. L'AFSSA est, dans le domaine de la nutrition, saisie sur de nombreux dossiers industriels d'autorisation : mise sur le marché, revendication d'une allégation, enrichissement d'un aliment de consommation courante, utilisation d'un nouvel aliment ou d'un nouvel ingrédient.
Dans le cadre de saisines ou d'autosaisine, l'unité nutrition a, par ailleurs, conduit au cours de la période 2001-2002 des réflexions scientifiques sur divers sujets de fond au sein de huit groupes de travail : consommation de sel et santé, intérêt nutritionnel et risques pour la santé de l'enrichissement des denrées alimentaires en acide gras omega 3, adjonction de pré et pro biotiques dans les préparations pour nourrissons, enrichissement des aliments courants en vitamines et minéraux, apports nutritionnels conseillés chez l'enfant sportif, compléments alimentaires à base de plantes, etc ... ».
* Son rôle et ses priorités
L'AFSSA est donc un acteur important du « Plan national nutrition santé » lancé fin janvier 2001 : elle a ainsi assuré la rédaction du fond scientifique des guides alimentaires grand public et professionnels et a préalablement participé à l'élaboration des différents objectifs prioritaires et axes stratégiques :
Les neuf objectifs prioritaires ont été ainsi définis :
- augmenter la consommation de fruits et légumes
- augmenter la consommation de calcium
- réduire la contribution moyenne des apports lipidiques totaux
- augmenter la consommation de glucides lents
- réduire l'apport d'alcool
- réduire la cholestérolémie moyenne
- réduire de 10 mm de mercure la pression artérielle systolique chez les adultes
- réduire la prévalence du surpoids et de l'obésité
- augmenter l'activité physique quotidienne
Parmi les cinq thématiques transversales retenues par l'Agence pour la période 2002-2005 au niveau de la recherche, « la composition des aliments et les risques nutritionnels » illustre aussi l'importance donnée à ce domaine.
La pondération des efforts et des actions en proportion des enjeux de santé publique, donc in fine en terme de vies, est pleinement justifiée par les réflexions et remarques maintes fois entendues de différents interlocuteurs. Abaisser un risque sanitaire de 1 pour 100.000 à 1 pour 1 million de cas peut exiger des études considérables (et incertaines) en impliquant à terme des mesures aux coûts socio-économiques énormes. Dans le même temps, des dérives nutritionnelles ou comportementales, qui n'ont rien d'hypothétique, peuvent être aisément identifiées pour être combattues et concernent des millions de personnes, l'obésité juvénile en étant l'exemple le plus frappant. Le directeur général de l'AFSSA ne manque d'ailleurs pas de rappeler cette nécessaire mise en perspective en rapprochant par exemple le nombre de victimes de listériose à celui des accidents de la route pendant un week-end.
Au passage, soulignant la gravité de la situation en matière d'obésité, on ne saurait trop insister sur la nécessité d'en faire aujourd'hui l'objectif premier de toute action dans le domaine nutritionnel : les données les plus récentes (enquête Obépi etc ...), heureusement largement répandues, suffisent comme argumentaires, mais l'effort actuellement consacré est tout à fait insuffisant, en France comme ailleurs en Europe. Les mesures récemment prises dans le cadre de la loi de santé publique et prévoyant notamment l'interdiction de la distribution de confiseries et boissons sucrées par automates dans les établissements d'enseignement relèvent de la même nécessité urgente, mais sont largement inadaptées à l'ampleur du fléau qui s'installe dans notre société.
Le besoin d'appui scientifique et d'action vers le grand public justifie donc pleinement les activités et efforts que l'AFSSA a déployés et déploie dans le domaine nutritionnel. Sans être aussi complexe que l'analyse des risques sanitaires, notamment face à des risques émergents, l'organisation des travaux dans ce domaine exige une certaine méthode et aussi un ciblage justifié de travaux qui peuvent rapidement prendre une ampleur incontrôlable. L'Agence intervient le plus souvent ici sur la base d'une auto-saisine. Elle a généralement recours à la constitution de groupes de travail qui sont le cadre logique d'une telle activité et ce, sans préjudice de l'appel à contribution d'experts d'autres cadres. En outre, elle peut être amenée à intervenir conjointement avec d'autres entités, au premier rang desquels l'InVS.
* Des avis parfois débattus
La formation, la composition et le fonctionnement des groupes de travail ont paru poser problème dans certains cas à nombre d'interlocuteurs. L'avis sur le sel dans l'alimentation en est une illustration. Un premier avis de l'AFSSA a été rendu le 13 juin 2000 ; ses conclusions étaient les suivantes :
« Le manque de certitudes scientifiques sur la consommation optimale de sel n'incite pas, à l'heure actuelle, à des recommandations définitives : une moyenne d'apports réels de 6 - 8 g de sel par jour permettrait de modifier la distribution des consommations de sel en France de telle sorte que la proportion des forts consommateurs (plus de 12 g/j) diminuerait.
Il n'apparaît pas nécessaire de lancer des campagnes publiques alarmistes et médiatiques sur le sel, au détriment d'autres enjeux de santé publique (tabac, alcool, obésité) et au risque de focaliser la nutrition sur un élément et non sur l'ensemble du régime.
Il est important d'associer l'industrie agroalimentaire dans cet objectif sanitaire dans la mesure où il semble qu'une grande partie du sel consommé proviendrait des produits industriels transformés. Un groupe de travail spécifique associant les industriels pourra être mis en place dans le cadre de l'AFSSA afin de poursuivre la réflexion sur le sujet. Ces travaux avec les industriels permettraient d'évaluer la faisabilité d'une réduction progressive de la teneur en sel des aliments transformés. Les conséquences des éventuelles mesures de réduction qui seront prises devront être évaluées après quelques années pour juger de leur pertinence.
Dans ce contexte, les études concernant la perception gustative qu'ont les consommateurs de la réduction des teneurs en sel des aliments devraient être complétées. Une communication devra accompagner toute mesure éventuelle de réduction des teneurs en sel des aliments transformés afin d'éviter leur sur-salage a posteriori. Enfin, il serait opportun que l'étiquetage renseigne davantage sur le contenu en sel des aliments : ces informations pratiques sont en effet indispensables aux personnes soumises à des régimes hyposodés (dits « sans sel).
Pour une meilleure estimation des apports sodés, il est important que la recherche soit développée à différents niveaux : mise à jour régulière des tables de composition des aliments concernant leur teneur en sel et détermination des apports sodés réels (natriurèse de 24 h) sur un échantillon représentatif de la population française.
Un recueil de données au long cours et régulier doit être mis en place, en relation avec les habitudes alimentaires, l'état nutritionnel, l'incidence de nouveaux cas de maladies cardiovasculaires et la mortalité cardiovasculaire. Au sein de ce système d'information, le sel prend sa place, au même titre que d'autres variables. Seul ce programme de surveillance rendra possible l'évaluation correcte des conséquences de l'environnement sur la population française en termes de maladies cardiovasculaires.
La réflexion sur le sodium va de pair avec celle sur le calcium et le potassium, l'objectif étant d'optimiser l'apport alimentaire de ces derniers, tout en maîtrisant celui du sodium ».
Le rapport sel de l'AFSSA (janvier 2002) lui-même présente la génèse des travaux de l'Agence à partir de « l'avis provisoire » précité :
« Suite à son avis provisoire sur le sel (rendu le 13 juin 2000) dans le prolongement des travaux menés dans le cadre de la rédaction des apports nutritionnels conseillés l'AFSSA s'est fixée une stratégie visant à :
1. proposer des actions ciblées sur la diminution des apports en sel ;
2. entreprendre des travaux pour connaître la consommation réelle individuelle de sel ;
3. entreprendre des recherches sur la teneur réelle en sel des aliments ;
4. faire un point des connaissances scientifiques sur les relations entre sel et santé.
Les différents axes de cette stratégie visent à fournir des données scientifiques validées qui servent de base à des recommandations de santé publique, contribuant notamment à atteindre l'objectif de réduction de l'hypertension artérielle fixé dans le cadre du programme national nutrition santé (PNNS).
L'avis de l'AFSSA du 13 juin 2000 annonce, dans sa conclusion, « la mise en place d'un groupe de travail spécifique, dont la mission sera d'évaluer la faisabilité d'une réduction progressive de la teneur en sel des aliments transformés ».
Ce groupe de travail a été formé en mars 2001, à la demande de l'AFSSA et de son directeur général, M. HIRSCH, associant l'ensemble des partenaires concernés : administrations, agences, acteurs économiques, associations de consommateurs et scientifiques ».
Les objectifs qui lui ont été fixés alors sont textuellement les suivants :
1. proposer des mesures à mettre en oeuvre pour respecter une distribution statistique de consommation de chlorure de sodium de 5 à 12 g/j ;
2. identifier les aliments vecteurs de l'essentiel de l'apport sodé alimentaire ;
3. proposer des recommandations effectives d'abaissement de la teneur en sodium de certains aliments vecteurs tout en respectant l'approche organoleptique, sécuritaire et technologique et réfléchir aux substituts potentiels du sel ;
4. effectuer des études de simulation de l'apport sodé de la population française ;
5. réfléchir sur les moyens de communication à adopter pour accompagner les mesures d'abaissement de la consommation de sodium.
L'objectif central des recommandations du groupe de travail qui constitue le rapport de janvier 2002 (100 pages) est une réduction de 20 % de l'apport alimentaire moyen de sel établie sur 5 ans (soit 4 % par an).
Le « principe général des recommandations » est ainsi explicité :
« Principe général des recommandations du groupe de travail :
*Stratégie
Pour modifier la distribution des consommations de sel ingéré dans la population française de manière à respecte une distribution statistique de consommation de sel comprise entre 5 et 12h/j, le groupe de travail propose de développer des actions générales dont l'impact se fera ressentir de façon préférentielle au niveau des grands consommateurs (c'est-à-dire les sujet consommant plus de 12 g/j). Les actions proposées doivent permettre de resserrer la distribution statistique des consommations de sel aux dépens des forts consommateurs.
Compte tenu de la définition des missions confiées au groupe de travail, il ne s'agit pas de proposer des recommandations visant à décaler globalement la distribution des consommations vers les consommations les plus basses par une simple translation (ce qui voudrait dire que l'effet des actions serait équivalent aussi bien pour les grands que les moyens ou petits consommateurs de sel). Il s'agit, en fait, de réduire proportionnellement plutôt la partie haute de la courbe de distribution statistique, correspondant aux grands consommateurs.
Le principe de la stratégie proposée dans ce rapport, pour tendre vers une distribution statistique de consommation de sel de 5 à 12 g/j, est donc de mettre en place des actions générales pour l'ensemble de la population devant avoir un impact plus net sur les grands consommateurs. Ces actions générales amèneront à diminuer raisonnablement la consommation moyenne de sel de la population tout en réduisant proportionnellement la fréquence des grands consommateurs de sel ( 12 g/j). Les actions de communication pourront particulièrement être ciblées de manière à avoir un impact plus fort sur les plus grands consommateurs. Les mesures générales de réduction de la teneur en sel de certains aliments vecteurs importants de sel ou la communication sur la limitation de leur consommation, agiront sur la consommation moyenne de sel, mais surtout sur les plus grands consommateurs de sel. Cet effet sur les forts consommateurs paraît assuré par le fait que les principaux aliments vecteurs de sel chez ces forts consommateurs (résultats similaires des études Inca et SU.VI.MAX) sont les mêmes que ceux de l'ensemble de la population, consommés en plus grandes quantités (pain/biscottes, charcuterie, soupes, plats composés, fromages et snacks). Les études de simulation présentées dans le chapitre 12 (page 68)) confirment la validité de cette hypothèse.
Les données disponibles sur la consommation de sodium dans la population française (enquêtes alimentaires, études spécifiques de natriurèse) suggèrent de façon cohérente des apports moyens de sel (provenant des aliments et ajouté) entre 9 et 10 g/j.
Compte tenu de ces estimations, il apparaît raisonnable de proposer des recommandations, en terme de santé publique, permettant de se fixer comme objectif une réduction de 20 % étalée sur 5 ans de l'apport moyen de sel, soit une réduction d'environ 4 % des apports sodés moyens par an. Les recommandations proposés au niveau de la réduction de la teneur en sel des aliments et de la communication visent à avoir un impact proportionnellement plus fort chez les grands consommateurs de sel.
La réduction de l'apport sodé moyen de 20 % étalée sur 5 ans (environ 4 % par an) :
• permettra d'atteindre en 5 ans, un
apport moyen de l'ordre de 7 à 8 g/j et d'avoir un impact en terme de
réduction de la prévalence des consommations supérieures
à 12 g/j.
• est suffisamment progressive pour rester
dans une limite acceptable pour le consommateur sur le plan
organoleptique,
• peut être atteinte par l'ensemble
des actions proposées dans ce rapport.
Cette approche est en cohérence avec l'avis de l'AFSSA concernant le sel qui, dans sa version provisoire, proposait dans ses conclusions « qu'une moyenne d'apports réels de 6-8 g de sel par jour, permettrait de modifier la distribution des consommations de sel en France, de telle sorte que la proportion des forts consommateurs (plus de 12 g/j) diminuerait ».
Les recommandations concernant le sel doivent s'intégrer dans le cadre de la politique nutritionnelle globale, visant à la prévention des grands problèmes de santé publique, qui sont à l'évidence des maladies multifactorielles.
Les recommandations ne visent pas à diaboliser le sel, mais à resituer le rôle de l'excès de consommation du sel parmi l'ensemble des facteurs nutritionnels de risque impliqués dans le déterminisme des maladies ».
Le groupe de travail est en effet « en cohérence » avec l'avis provisoire de l'AFSSA de juin 2000. On peut se demander si l'ensemble de cette activité qui s'est déroulée de juin 2000 à janvier 2002 n'avait pas pour objectif principal de valider ces recommandations préalables, ce qui est tout à fait normal, mais aussi des recommandations qui avaient de toute façon été décidées. Les conditions dans lesquelles le groupe de travail a fonctionné semblent parfois avoir été difficiles. Le développement de campagnes médiatiques qui pouvaient paraître synchronisées avec le calendrier du groupe a été remarqué d'autant que certains de ceux qui y apparaissaient étaient eux-mêmes les militants les plus convaincus d'une des thèses en présence au sein du groupe de travail et ce, d'une façon assez éloignée des rigueurs de la démarche scientifique.
Dès lors, il n'est pas étonnant que plusieurs « positions minoritaires » soient apparues sous forme de résumés à la suite du rapport.
-- celle du Dr Tilman B. Drueke (Directeur de recherche - unité 507 de l'INSERM et service de néphrologie, hôpital Necker-Paris :
1. « Je ne donne pas ma caution à la recommandation de réduire progressivement la consommation de sel chez l'ensemble de la population française de 4 % par an et ce pendant cinq années consécutives, en utilisant l'argument qu'il n'y a guère d'autre moyen pour parvenir à une réduction des ingesta chez les sujets qui consomment des quantités excessives, c'est-à-dire plus de 12 g de sel par jour. Cette recommandation est d'autant plus surprenante qu'on ne connaît pas de façon précise le pourcentage de la population française qui consomme plus de 12 g par jour. De plus, il n'est pas démontré qu'une telle mesure générale améliore la santé de la population, à l'exception peut-être des sujets obèses. Pour ces derniers, mieux vaut maigrir en augmentant l'exercice physique et en mangeant de façon plus équilibrée que consommer moins de sel.
2. L'imposition à l'ensemble de la population d'une réduction de la consommation de sel de 20 % implique un régime pratiquement désodé pour la frange de la population qui se trouve dans le quartile inférieur de la distribution des ingesta sodés. Je ne donne pas un avis favorable à ce type de mesure ».
-- Celle de M. Léon Guéguen, directeur de recherches honoraire INRA
Avertissement pour le CES « Nutrition humaine » :
« Le présent commentaire ne constitue nullement une critique du fonctionnement du groupe « sel » ni une remise en cause de la qualité et de l'importance du travail effectué. Le mérite du Président Serge Hercberg dans une situation aussi conflictuelle doit être souligné ! Ce commentaire vise seulement à exprimer un point de vue largement évoqué au cours des débats et qui n'apparaît pas bien dans le rapport.
Avis exprimé :
« Sans remettre en cause l'importance et la qualité du travail effectué par le groupe, ni l'intérêt pour la santé publique d'une réduction d'apports sodés toujours très supérieurs aux besoins, L. Guégen émet des doutes sur l'efficacité de certaines actions envisagées. Il préfère les actions incitatives aux mesures réglementaires et considère que le « libre choix éclairé et responsable » du consommateur, qu'il faut améliorer par l'éducation, l'information (dont l'étiquetage), le conseil diététique et médical, doit être privilégié. Il doute de la portée réelle d'une faible réduction des teneurs en sodium de quelques aliments imposée à l'ensemble de la population et considère que, pour bien atteindre la cible visée, des recommandations plus fortes devraient concerner les gros consommateurs de sel à risque avéré et facilement identifiables (hypertension, obésité, insuffisance cardiaque ou rénale). »
-- Celle du Pr Pierre Louisot - Laboratoire de biochimie INSERM Unité 189 CNRS - Faculté de médecine Lyon-Sud :
« 1. Le Président et les membres du groupe de travail ont fait un effort méritoire et important de documentation, sur un sujet complexe, dont il convient incontestablement de les remercier.
2. Si j'apprécie comme il se doit cet effort, à l'opposé, je me désolidarise fondamentalement de la recommandation visant à réduire pour l'ensemble de la population la consommation de sel de 20 % en cinq ans. Les raisons en sont les suivantes.
3. Malgré des tempêtes médiatiques anciennes ou récentes, il n'est nullement prouvé, sur des bases scientifiques incontestables, que la réduction de la consommation de sel soit en relation de cause à effet directe avec la prévention de l'hypertension artérielle et des maladies cardiovasculaires chez l'homme pour la population courante. Le sujet demeure très largement controversé.
4. Les gros consommateurs de sel sont généralement des gros consommateurs de tout. Ils ne représentent probablement qu'environ 20 % de la population, alors que 80 % de celle-ci se situent dans des normes de consommation en sel tout à fait raisonnables. Il est illogique de s'attaquer à l'ensemble de la population, surtout par voie réglementaire, par une mesure qui est loin d'être anodine, alors qu'une fraction minoritaire de cette population est seule concernée.
5. Tous les spécialistes savent que le métabolisme chez l'homme est un ensemble complexe, finement régulé, et d'ailleurs équilibré de manière très différente dans les diverses régions du monde. C'est un domaine où le multifactoriel est de règle. La tentation de normaliser ce métabolisme par une action sur un seul paramètre considéré comme coupable, est grande. L'exemple des obèses, qui se refusent à modifier l'ensemble de leur comportement et espèrent que la correction d'un seul paramètre améliorera leur sort, est tristement d'actualité. Encourager la population à la correction nutritionnelle monofactorielle n'est pas adapté à la situation, alors que tout devrait au contraire converger vers des attitudes d'ensemble portant aussi bien sur l'alimentation que sur l'activité physique.
6. Enfin, l'habitude du sel à concentration raisonnable induit normalement des comportements nutritionnels au quotidien qui pourraient être significativement déséquilibrés par une modification sensible des qualités organoleptiques des nouveaux produits proposés. Personne ne peut sous-estimer d'éventuels effets pervers de cette mesure : la restriction de la consommation de pain - que l'on recommande par ailleurs d'augmenter dans l'alimentation - étant le premier exemple qui vient à l'esprit, mais il y en aura sans doute bien d'autres, y compris à conséquences économiques, touchant les fromages ou la charcuterie, lesquels font partie intégrante et bien comprise de l'équilibre nutritionnel des Français ».
Bien que le souci nutritionnel soit au coeur des compétences de l'AFSSA, on touche là, avec cet « avis provisoire » puis le rapport qui suit, les limites de la compétence de l'Agence. Elle peut subrepticement devenir un enjeu dans des « luttes militantes » où des intérêts de tous ordres peuvent l'amener à être quelque peu manipulée et à s'éloigner de l'appréciation du risque, fut-il nutritionnel. Le rapport sur l'agriculture biologique semble, dans une autre tonalité, avoir posé aussi quelques problèmes, la pratique du « rapport intermédiaire » introduisant un élément de trouble qui est peut-être à éviter. Lorsque le directeur général de l'AFSSA évoquait lors de l'audition de mai 2000 à la commission des affaires sociales du Sénat, l'importance à accorder aux travaux de fond, on ne peut qu'être d'accord avec son analyse, mais un peu moins avec l'exemple choisi :
« La deuxième perspective est la nécessité de pouvoir maintenir une capacité à développer des travaux approfondis. Si nous voulons bien guider l'action des pouvoirs publics, il faut pouvoir lancer des réflexions de fond mettant en perspective les risques, les vulnérabilités, les enjeux.
Il n'y a aujourd'hui aucune situation de crise dans certains domaines où nous pensons qu'il faut quand même mettre à plat un certain nombre de sujets : c'est ce que nous faisons par exemple pour l'agriculture biologique, en nous disant que la situation est suffisamment précoce pour que l'on soit capable de faire une analyse objective. Si nous n'avions pas les moyens de mener ces travaux de fond, si tous les moyens étaient obérés par la réponse aux crises, nous serions revenus à la case départ ».
En conclusion, il reste à préciser des règles pour ce type de tâche qui est essentielle afin notamment de cadrer les choix d'auto-saisine et les conditions de fonctionnement des groupes de travail.
II. AU-DELÀ DE LA REUSSITE GLOBALE, DES AJUSTEMENTS NECESSAIRES
La mise en oeuvre de la loi du 1 er juillet 1998 dans le domaine de la sécurité des aliments a nécessairement été une réalisation dans la durée car contrairement à l'AFSSAPS qui succédait à l'Agence du médicament où à l'InVs qui prenait la suite du réseau national de santé, l'AFSSA constituait un élément nouveau, même si l'intégration du CNEVA avec ses laboratoires et le pôle du médicament vétérinaire de Fougères lui donnaient au départ des éléments fonctionnels nécessaires comme le législateur l'avait d'ailleurs voulu.
Cette notion de durée va de pair avec le pragmatisme qu'exigeait l'ajustement des nouveaux mécanismes d'une sécurité sanitaire faisant intervenir d'autres acteurs que l'AFSSA. Cette observation justifie la nécessité de vérifier tout d'abord le principe de base sur lequel la sécurité sanitaire des aliments est fondé : la séparation de l'évaluation du risque et de la gestion du risque. Cette dernière notion pourrait être mieux exprimée par le terme de « décision » car c'est là l'élément fondamental de cette phase du processus. Tout en gardant cette nuance à l'esprit, on en restera à la distinction classique.
Cette vérification débouchera sur une confirmation du rôle de la plupart des autres acteurs, c'est-à-dire essentiellement les trois tutelles que l'on pourrait peut être maintenant qualifier plus exactement de « partenaires ministériels ». La DGCCRF (Ministère de l'économie, des finances et de l'industrie), la DGAL (Ministère de l'Agriculture, de l'alimentation, de la pêche et des affaires rurales), la DGS (Ministère de la santé et de la protection sociale).
S'agissant des relations entre ces différents acteurs, la confirmation des principes retenus et des compétences n'excluent pas, bien au contraire, des ajustements pour deux séries de raisons au moins. D'une part, les mécanismes d'information et de coopération prévus par les nouvelles dispositions ne fonctionnent pas toujours comme ils le devraient. D'autre part, des évolutions exigent que l'action publique soit sans cesse adaptée pour faire face à des exigences ou des risques nouveaux.
Des ajustements sont aussi nécessaires dans l'organisation interne dans l'activité fondamentale de l'AFSSA elle-même : l'évaluation du risque à travers les saisines qu'elle a à traiter.
Des lacunes de l'architecture générale de la sécurité de la sphère alimentaire exigent qu'il soit procédé à des ajouts et des précisions à la construction de 1998, dans des domaines qui n'ont sans doute pas à l'époque été perçus comme centraux, mais qui posent de réels problèmes : il s'agit notamment du domaine végétal avec les produits phytosanitaires.
Des mises au point ou d'autres clarifications sont nécessaires dans deux aspects très différents de l'action de l'Etat. Il s'agit d'une part du rôle des laboratoires qui appelle une réflexion d'ensemble, quel que soit leur ancrage administratif et, d'autre part, du « process européen » de la sécurité des aliments ; si la clarification à réaliser ne fait pas de doute, les moyens d'y procéder ne dépendent évidemment pas de la seule agence française, ni même du seul Etat-membre qu'est la France.
2.1. La séparation entre l'évaluation et la gestion : un principe vérifié
La vérification de ce principe est d'autant plus nécessaire qu'il a été récemment remis en cause alors qu'un consensus semblait s'être peu à peu établi, puis renforcé.
2.1.1. Un principe non universel
Indépendamment de toute nouvelle approche, on peut considérer que cette séparation ne se retrouve pas dans d'autres Etats, ou en tout cas pas organisée d'une manière aussi nette.
Ainsi en Grande-Bretagne, la création de la Food Standards Agency instaurée par la réforme de 1999 (Food Standards Act) a eu pour objet de restructurer complètement l'organisation de la sécurité sanitaire des aliments face à une « nécessité de reconquête de l'opinion » selon les termes du directeur exécutif de la FSA lui-même ; la confiance du public avait en effet été très atteinte à la suite de la crise de l'ESB et des épidémies de salmonellose. Pour reprendre les termes de Sir John Krebs, président de la FSA, la loi de 1999 a créé un « département gouvernemental, (c'est-à-dire un ministère sans ministre (...), la fonction de protection des consommateurs étant ainsi séparée de l'influence directe des ministres ». La FSA a repris les fonctions du ministère de l'agriculture et de la santé dans ces domaines et acquis de nouveaux pouvoirs qui relevaient précédemment directement du gouvernement ; en outre, elle participe concrètement à l'élaboration des actes normatifs, y compris les textes européens et internationaux et a le pouvoir de proposer au Parlement l'adoption de mesures susceptibles d'améliorer la sécurité dans ce domaine.
Enfin, et c'est évidemment essentiel, les avis rendus par la FSA en matière de sécurité des aliments suite à une saisine doivent être suivis par le Gouvernement sauf à en donner la motivation publiquement. Le président de la FSA résume ainsi le positionnement de l'Agence : « l'objectif de notre institution est de protéger la santé du public et les intérêts des consommateurs en matière alimentaire. Notre mandat va donc bien au-delà de la sécurité alimentaire ».
Le dispositif retenu en Grande-Bretagne doit donc être remis dans le contexte de la crise de confiance majeure des années 90. On est ainsi passé d'un fonctionnement confidentiel des instances administratives en charge de l'évaluation et de la gestion à une exposition publique des principales phases d'action de la nouvelle FSA. En outre, les spécificités permanentes de l'organisation du pays doivent être présentes à l'esprit ; ce sont les autorités locales qui sont chargées de l'application des lois et règlements sur le terrain ; par ailleurs, les analyses nécessaires aux contrôles sont presque toutes externalisées : il n'y a pas en Grande-Bretagne l'équivalent des laboratoires dont on dispose en France à l'AFSSA.
Sans aller plus avant dans l'analyse comparative entre la France et la Grande-Bretagne, il apparaît clairement, comme c'est souvent le cas, que la structuration des mécanismes décisionnels est très liée à une situation institutionnelle et historique spécifique, elle-même marquée par les conséquences d'une crise majeure. En conséquence la transposition n'aurait guère de sens.
D'autres pays d'Europe connaissent d'ailleurs une absence de séparation dans le domaine alimentaire entre l'évaluation et la gestion du risque. C'est ainsi le cas de la Belgique et du Danemark.
2.1.2. Des remises en cause récentes
L'évolution des situations, le « retour d'expérience » ou tout simplement l'observation et le pragmatisme pourraient légitimement justifier une remise en cause du principe de séparation auquel on ne saurait conférer un « caractère théologique ». Deux démarches en ce sens peuvent être signalées : la première apparaît à travers une « auto-évaluation » à laquelle il a été procédé au deuxième trimestre 2003 à la demande de l'AFSSA elle-même, la seconde est une prise de position énoncée en novembre 2003 par le Pr. Lucien Abenhaïm, ancien Directeur général de la santé, dans son livre « Canicules ».
La « démarche d'auto-évaluation de l'AFSSA »
Une rapide présentation et explication de texte sont ici nécessaires.
Cette démarche d'auto-évaluation de l'AFSSA est ainsi présentée par son directeur général (document daté du 25 août 2003).
« L'AFSSA a souhaité réaliser un travail d'évaluation interne de son activité, trois ans après sa création. Cette démarche décidée en Comité de direction s'inscrivait dans un double contexte, celui de la mise en oeuvre des missions d'inspection ministérielle et parlementaire liées à l'évaluation de la loi de 1998 et celui de la fin du premier mandat des comités d'experts spécialisés de l'AFSSA en août 2002.
Les objectifs principaux étaient de faire le point sur les réalisations de l'AFSSA, la manière dont elle avait rempli ses missions mais également les manques, les difficultés et les domaines à renforcer afin de préparer une nouvelle étape dans le développement de l'agence après les phases de création et de construction.
Le choix a été fait de faire appel à une équipe externe, reconnue pour son expérience en évaluation des systèmes ou politiques publiques, appartenant à un organisme public, le CNRS avec un travail conduit en plusieurs étapes comprenant une phase externe complémentaire de la réflexion conduite en interne par les équipes de l'agence notamment au sein de la Derns en vue du renouvellement des comités d'experts ».
La note de synthèse d'accompagnement du processus d'auto-évaluation, selon les termes de cette présentation « portait sur le diagnostic que cette équipe spécialisée dans la sociologie des organisations faisait des premières années de l'agence au regard de ses missions et devait ouvrir des perspectives et faire des recommandations pour l'avenir ».
La méthodologie adoptée mais surtout les conditions dans lesquelles cette « auto-évaluation » a été menée ne nous paraissent pas de nature à tirer des enseignements substantiels dans le cadre de l'évaluation à laquelle nous sommes amenés à procéder même si le recueil d'opinions individuelles et de rappels de certains éléments factuels, le plus souvent déjà connus, ne manque pas d'intérêt. L'absence de recul, inévitablement lié à ce genre d'exercice, limite déjà l'apport attendu, et le non respect de règles classiques d'analyse sociologique oblitère nécessairement le résultat. Le fait que deux des trois tutelles ministérielles aient refusé de participer à cet exercice en rend les enseignements attendus d'autant plus limités. Des appréciations sévères sur ces travaux ont d'ailleurs été formulées, y compris par des connaisseurs ou professionnels de la sociologie des organisations.
Concernant la « note de synthèse », celle-ci procède en effet à des « diagnostics » et « ouvre des perspectives et fait des recommandations ». On y trouve ainsi une observation selon laquelle « Au plan législatif, en somme, la situation semble claire. Si l'agence n'a pas de véritable pouvoir de gestion du risque, elle peut se considérer comme chargée d'une mission d'évaluation de cette gestion par les autorités compétentes » . Les suggestions implicites que comporte cette rédaction méritent déjà d'être remarquées car elles traduisent assez clairement la tentation d'aller nettement au-delà de l'évaluation du risque pour ce qui est du domaine de compétence de l'agence. Très logiquement, ce que l'on trouve dans la conclusion en est le prolongement : la situation diagnostiquée de l'agence comporterait un risque : « celui de maintenir l'agence dans le créneau étroit de producteur d'avis scientifiques et de gestionnaire déléguée d'un potentiel de recherche et d'appui scientifique et technique, créneau qui semble convenir à la fois à ses administrations de tutelle et aux professionnels, mais qui apporte une « fausse » garantie en matière de maîtrise des risques ». (...)
« La question que doit se poser la direction générale est donc la suivante : l'AFSSA peut-elle se contenter d'occuper ce créneau tout en remplissant son rôle de protection de la santé publique ? (...).
« En revanche, si la réponse à cette question est négative, la perspective change et un autre axe d'action prioritaire apparaît : il s'agit alors de rechercher un autre positionnement de l'agence dans le champ de l'évaluation et de la gestion de la sécurité sanitaire des aliments. Une telle recherche devra se faire dans deux directions principalement qui impliquent
- de reconstruire d'autres rapports avec les représentants et les acteurs du secteur privé, en instaurant des relations plus directes reposant sur une forme de réciprocité ;
- de clarifier et surtout de rééquilibrer les relations de l'agence avec ses tutelles ».
(...)
« Cette clarification et ce rééquilibrage renvoient enfin, et par-dessus tout, à la réalisation du rôle que la loi confère à l'Agence dans l'évaluation des activités de tous les acteurs concernés par la gestion du risque alimentaire (...).
Si l'AFSSA souhaite renforcer son influence dans l'évaluation et la gestion de la sécurité sanitaire des aliments, tout en diminuant la vulnérabilité inhérente à la situation qu'elle occupe aujourd'hui, il lui revient de repenser dans un sens plus ouvert et plus direct ses relations avec les acteurs privés de la chaîne agroalimentaire et, parallèlement, à rééquilibrer les relations avec ses tutelles dans les directions esquissées ci-dessus ».
On distingue assez difficilement ce que ce « rééquilibrage », dont la nécessité est plusieurs fois soulignée, laisserait dans la gestion du risque ou à la décision aux directions ministérielles de tutelle. Les « relations directes avec le secteur privé reposant sur une forme de réciprocité » reviendraient à une situation que l'on a précisément voulu supprimer pour éviter la confusion des genres. L'ensemble des fonctions serait ainsi regroupé, pour ne pas dire confondu, dans le périmètre de l'AFSSA, seule compétente en matière de sécurité des aliments.
Dans une perspective moins transparente, c'est dans le même sens que s'orientent les propositions du Pr. L. Abenhaïm dans son livre « Canicules » 24 ( * ) . .
Consacrant un chapitre (« la sécurité sanitaire inachevée ») entier à l'organisation de la sécurité sanitaire et à l'architecture constituée par les agences à partir de la loi de 1998, il précise d'emblée : « la faiblesse de la réforme majeure de la sécurité sanitaire qui a eu lieu ces dix dernières années est qu'elle reste inachevée ».
Présentant l'Etablissement français du sang, l'Institut de veille sanitaire et l'AFSSAPS comme des réussites accomplies de la loi de 1998 et des réformes qui l'avaient précédée, l'auteur s'appuie précisément sur l'exemple de cette dernière où l'évaluation et la gestion du risque sont dans une même main pour estimer que « le secteur alimentaire est à la traîne » , selon l'intitulé même du paragraphe où il détaille son point de vue.
Les remarques et l'analyse précises des enchevêtrements de compétences décrivent assez bien les difficultés rencontrées, les risques de retard et la position particulièrement inconfortable de la D.G.S. ; l'exemple des importations est d'ailleurs particulièrement intéressant : il cite à titre d'illustration une infection de poulets importés des Pays-Bas par un virus de grippe potentiellement très dangereux pour l'homme en mai 2003 dont il a été informé par le ministère de l'agriculture avec plusieurs semaines de retard. L'illustration la plus marquante des graves difficultés systémiques avec le ministère de l'agriculture est celle des épidémies ou épisodes de listeria en 1993 et 1997. Les preuves bactériologiques se faisant inévitablement attendre, il convenait, en effet, à partir des constatations épidémiologiques faites par l'InVS de prendre des mesures conservatoires d'urgence. Et ce n'est qu'après de longues tractations à l'occasion de cette crise à l'automne 1999 qu'un protocole sur la listériose a pu être adopté et mis en oeuvre avec des résultats concrets.
Ces exemples et de nombreux autres illustrent d'une part les extrêmes difficultés et acuité des crises auxquelles la D.G.S. et le Ministère de la santé ont à faire face, et d'autre part un comportement discutable du Ministère de l'agriculture fondé sur des réflexes et des pratiques inadaptées face à l'évolution des esprits (exigence de transparence) et des techniques ou des situations (rapidité et diversité de la distribution, multiplicité de produits et de circuits).
En fait, l'AFSSA (ou l'InVs, en tant que tel), n'est pas en cause ; la conclusion que tire le Pr. L. Abenhaïm de ces faits sur la réunification de l'autorité évaluatrice du risque et celle investie du pouvoir de décision, n'en résulte pas réellement. La majorité des crises citées à l'appui de cette thèse sont antérieures à la création de l'AFSSA ou directement concomitantes à sa mise en place (listériose - automne 1999).
En fait, ce qui apparaît clairement au début de ce paragraphe intitulé « La position difficile des agences sanitaires » est que l'auteur n'a pas admis le partage des tâches et l'équilibre institués par la loi de 1998 dans le domaine alimentaire : il conclut d'ailleurs sans ambages : « La mission laissée à l'AFSSA en matière de sécurité sanitaire se limite donc à l'évaluation des risques et à la vérification de la conformité des décisions générales des ministères - leurs arrêtés - aux principes découlant de cette évaluation. Sans qu'elle ait les moyens de vérifier si ces décisions sont bien appliquées, ni même la capacité de les définir de façon opérationnelle. C'est extrêmement regrettable ».
Cette présentation restrictive du rôle de l'AFSSA est exagérée. Ses travaux, ses activités et la conception que son directeur général affiche du rôle de l'AFSSA malmènent d'ailleurs quelque peu cette analyse et cette conclusion lapidaire, même si, et c'est un problème d'application sur le terrain et non de principe, l'association de l'agence aux activités de contrôle sur le terrain peut soulever de réelles difficultés qui doivent être résolues rapidement. C'est d'ailleurs la résolution de ces difficultés qui permettra précisément de supprimer ces situations où le décideur est confronté au dilemme du « tout ou rien » qui est effectivement celui du ministère de l'agriculture ou de la santé.
Rappelons enfin que le choix de la séparation entre l'évaluateur et le gestionnaire du risque dans le domaine alimentaire n'est pas une exception française et qu'il est même l'un des principes d'organisation préconisé dans le cadre du Codex alimentarius.
2.1.3. Une confirmation souhaitable
Les opinions paradoxales ont au moins un mérite : celui d'obliger à remettre en cause une approche qui s'impose comme une évidence « incontournable » et définitive. Cela posé, l'argumentaire en faveur de la suppression de la séparation entre l'évaluation et la gestion du risque dans le domaine alimentaire paraît bien mince, circonstanciel et attaché à la forme plutôt qu'à la réalité des problèmes et des évolutions. En outre, la proposition de création d'un « grand ministère des risques » (Pr. L. Abenhaïm) dépasse largement le cadre de l'AFSSA pour atteindre un niveau de généralités qu'on abordera par ailleurs si tant est que cette philosophie puisse avoir des prolongements opérationnels.
Force est de constater qu'à l'exception des deux remises en cause précitées, la totalité des nombreuses et fort diverses personnalités interrogés ont d'emblée confirmé la nécessité de ce principe de séparation et ce, quels que soient leur situation, leur rôle ou leur absence de rôle, dans l'architecture institutionnelle mise en place depuis 1998.
Parmi les praticiens les plus expérimentés de la question, M. Christian Babusiaux, qui a exercé longuement les fonctions de directeur général de la concurrence, de la consommation et de la répression des fraudes du ministère de l'économie et des finances, a souligné qu'il avait voulu cette séparation dès le départ et qu'à l'usage son bien-fondé se confirmait sans ambages. Si la rédaction de certains avis de l'AFSSA par les préconisations qu'ils contenaient a pu susciter des difficultés (cas du mouton lors d'une épidémie de « tremblante » alors que l'essentiel de la consommation était alors issu d'importations), le principe est cependant justifié et opérationnel, ce qui n'exclut pas des ajustements dans certains types de situations.
Du côté des professionnels de la distribution, M. Jérôme Bédier (Président de la fédération des entreprises du commerce et de la distribution) a souligné que l'on tenait beaucoup à cette séparation qui était un acquis pour tout le monde en terme de sécurité.
Parmi les argumentations excluant le regroupement au sein de l'AFSSA, au-delà de l'évaluation du risque de l'ensemble des autres fonctions et notamment ce qu'on appelle la gestion du risque, la ministre en charge de la consommation, donc de la DGCCRF, Madame Marylise Lebranchu, avait indiqué en mai 2000, lors de la journée d'auditions publiques organisée par la commission des affaires sociales du Sénat sur la mise en oeuvre de la loi du 1 er juillet 1998 : « Avoir une seule entité impliquerait une supra structure avec à l'intérieur tous les laboratoires, le tout en étant au service de l'évaluation du risque. Je reste persuadée qu'il faut garder des outils au service de la gestion du risque. L'Agence n'a pas comme vocation de contrôler. Elle perdrait son temps et son efficacité. En effet, le souci, que vous avez bien développé, était d'avoir un outil structurel institutionnel garanti transparent travaillant en permanence sur l'évaluation des risques potentiels. Laissons la gestion aux administrations de telle manière que l'Agence ne soit pas encombrée de missions de contrôle et ne fasse plus de travail d'évaluation aussi souvent qu'elle le pourrait.
Par ailleurs, il me semble que l'Agence n'aura jamais tous les laboratoires sur toutes les compétences dans l'avenir. Qu'elle ait le droit, en tant que de besoin, d'avoir à sa disposition ces laboratoires et que ceux-ci soient obligés de lui répondre sur toutes les évaluations qu'elle demande, y compris les laboratoires universitaires - des universités travaillent sur de la recherche fondamentale qui, à un moment donné, peuvent être utiles à l'Agence - est un fonctionnement qui me paraît devoir être poursuivi ; sinon, l'Agence manquera de moyens ».
Cette analyse faite un an après la création de l'AFSSA garde toute sa valeur et illustre clairement les risques d'un tel regroupement. Mais le corollaire de la répartition des fonctions ainsi retenue apparaît aussi clairement : l'accès aux laboratoires extérieurs à l'Agence et l'obligation pour eux de répondre à ses questions doivent être assurés. Sur ce point, précisément, un effort réel reste à faire pour que l'équilibre d'ensemble instauré en 1998, et donc l'efficacité recherchée, soit atteint.
Enfin, s'agissant du pouvoir de décision dans le domaine alimentaire, on voit mal celui-ci exercé par l'autorité administrative qui aurait procédé préalablement à l'évaluation : les critiques sur la confusion des fonctions formulées avant 1998 retrouveraient alors toute leur justification.
Ce point essentiel du dispositif est aujourd'hui peu controversé. Au total, cette distinction entre évaluation et gestion du risque constitue un progrès marquant et si des aménagements dans la communication des avis peuvent apparaître nécessaires dans certains cas, l'architecture d'ensemble n'est pas remise en cause.
2.1.4. Une communication mieux cadrée
Dans un domaine de la vie quotidienne où la sensibilité de la population est très marquée et s'est même renforcée avec l'enchaînement de crises ou d'épisodes accidentels fort médiatisés, la communication est naturellement un élément essentiel. Non seulement elle a été longtemps insuffisante, voire inexistante, mais encore les difficultés qu'elle recélait constituaient le problème principal. La nouvelle architecture instituée par la loi de 1998 a d'ailleurs eu notamment pour objet de répondre à ce besoin de communication en fixant les rôles de chacun : - AFSSA, en tant qu'évaluateur du risque - tutelles ; l'Institut de veille sanitaire joue un rôle essentiel en amont (listérioses par exemple).
2.1.4.1. Le fondement juridique
« En vue de l'accomplissement de ses missions, l'AFSSA ... rend publics ses avis et recommandations », (art. L 1323-2 du code de la santé publique)
Les prérogatives de communication de l'Agence à cet égard ne font aucun doute comme cet extrait du texte fondateur de la loi de 1998 le montre. Mais si le domaine que traitent les avis de l'Agence est d'ordre scientifique, la communication, élément essentiel vis-à-vis de la population, n'est pas unanime ni même univoque ; elle semble avoir varié avec le temps.
Sur le principe, personne ne conteste le droit et même le devoir de l'Agence de communiquer sur ses travaux et en particulier sur les avis qu'elle rend : la prérogative précitée est conçue comme une pratique courante destinée notamment à mettre un terme aux silences du passé. Dès la mise en place de l'Agence, son directeur général a donné à la communication une vigueur, une rapidité et une efficacité appréciables. Le protocole signé en 1999 à la suite d'une épidémie de listériose avec les trois ministères de tutelle a précisé les conditions dans lesquelles l'information leur est transmise.
La mise en oeuvre semble être spécialement appréciée des journalistes, un peu moins de certaines administrations de tutelle. La qualité de l'information n'est pas en cause. Elle est reconnue et d'ailleurs procède normalement des comités d'experts qui réalisent l'évaluation elle-même. Sa lisibilité pour l'essentiel est appréciée, même si dans certains cas l'aide d'un scientifique paraît nécessaire pour décrypter sans risque d'erreur le texte de l'avis.
Le caractère centralisé, « cadré » de l'information ainsi donnée par l'Agence est parfois critiqué au nom d'une diversité scientifique d'analyse que l'on ne retrouverait pas dans l'avis. Il semble qu'il y ait là une confusion entre la diversité de la recherche et la nécessité d'une évaluation à des fins sanitaires. On imagine sans peine les critiques, très justifiées, de textes qui rendraient compte du balancement entre les différentes approches possibles et les gradations dans les appréciations de la situation.
2.1.4.2. Les reproches multiples
Les reproches sont par contre plus nombreux et, semble-t-il, plus consistants en ce qui concerne la rédaction des avis et la temporalité dans la communication.
Ces remarques concernent essentiellement les périodes de crises, les dossiers sensibles ESB, notamment les farines animales, tremblante du mouton par exemple. Cet aspect qui en fait illustre les limites du succès de l'AFSSA a été traité dans la première partie (l'évaluation globale).
La rédaction de l'avis : le contenu même de l'avis ne laisserait guère de marge de choix aux ministères chargés de la décision en aval : dans le cas des farines animales par exemple.
La multiplicité des avis successifs sur la même question contribue indiscutablement à « faire monter la pression » dans l'opinion et ce, sans réelle justification scientifique. Certes, la mise en garde rituelle et fondée le plus souvent « dans l'état actuel de nos connaissances scientifiques et de nos informations » peut fournir une telle justification, mais cela n'autorise pas pour autant une attitude qui peut aller à l'encontre de l'objectif de transparence et de sérénité affiché.
L'une des critiques la plus tangible est celle portant sur le délai insuffisant laissé aux autorités de tutelle pour prendre connaissance de l'avis avant sa diffusion dans le public.
Sur les questions les plus sensibles, cela a souvent été le cas. Il n'appartient évidemment pas au Parlement de préciser les règles de fonctionnement à cet égard. Mais on peut souhaiter que tout soit fait pour ne pas donner prise à ces critiques ; sinon le soupçon de la volonté de bousculer l'équilibre instauré par la loi entre l'Agence et les autorités ministérielle prendrait une consistance certaine, ce qui pourrait entraîner des attitudes regrettables par rapport aux progrès acquis en matière de transparence.
D'une manière générale, une certaine ardeur excessive de communication qu'a manifestée l'Agence pendant ses trois ou quatre premières années d'existence est retombée, ce qui apparaît positif.
Au-delà de règles de bon fonctionnement, de bon voisinage et de bon sens qui ne paraissent pas trop difficiles à trouver et à respecter, un ou deux principes simples peuvent être retenus : bannir le principe de la communication immédiate dès lors qu'il n'y a pas urgence : la capacité physique à tout présenter sur Internet ne justifie pas l'affichage automatique ; dans le même esprit une périodicité de publication régulière faciliterait la résolution de ce problème réel.
La conception de l'avis lui-même a fait l'objet de remarques de différents horizons. Les avis de l'Agence sont signés du directeur général personnellement engagé scientifiquement et juridiquement. Mais le texte de l'avis du comité d'expert n'est pas publié, ce que certains regrettent. Il reste que le directeur général peut disposer d'une marge d'appréciation. La lisibilité, la nécessité du caractère opératoire de l'avis rendu peuvent justifier ce décalage quand il existe. Toutefois, pour limiter la portée de cette difficulté, la mention sur l'avis de l'AFSSA de l'absence d'objection du président du comité (« nihil obstat ») serait à envisager.
De même, l'absence de coordination entre les avis des comités d'experts lorsque plusieurs d'entre eux sont concernés par une saisine devrait aisément être surmontée car elle est injustifiable. Par ailleurs, il serait souhaitable de faire fonctionner le Conseil scientifique en formation restreinte, ce qui est prévu par les textes (art. R794-22), cela permettrait de faciliter la cohérence entre les avis de dix comités d'experts spécialisés.
2.2. La coopération entre évaluateurs et gestionnaires : des progrès à réaliser
La fonction régalienne d'assurance de la sécurité des aliments dont l'Etat a la charge implique que les différents acteurs qui y participent oeuvrent d'une manière concertée, d'autant que la distinction qui a été établie entre agence chargée de l'évaluation du risque et services ministériels chargés de la gestion du risque et du contrôle (l'autorité ministérielle gardant le pouvoir de décision) est fonctionnelle et vise précisément à atteindre la meilleure efficacité. Les textes fondateurs ne laissent d'ailleurs aucun doute à ce sujet :
« Pour l'accomplissement de ses missions, les laboratoires des services de l'Etat chargés du contrôle de la sécurité sanitaire des aliments et ceux qui leur sont rattachés sont mis à disposition de l'Agence en tant que de besoin » (art. 1323-1, 4 ème alinéa du code de la santé publique) - L'art. R 794-2 de ce même code précise ce rôle et cette situation, notamment dans le domaine vétérinaire - L'appui scientifique et technique que l'Agence doit de son côté assurer au ministère de l'agriculture et aux autres ministères est également précisé.
Les différents avis et opinions recueillis conduisent à constater que cette coopération ne se vérifie pas dans le fonctionnement quotidien avec, sans doute, des variations selon les administrations de tutelle.
Une première remarque permet de relier cette observation à celles faites (cf. supra) sur la séparation entre l'évaluation et la gestion du risque. L'administration la plus réticente à la création de l'AFSSA, à savoir le ministère de l'agriculture, considère que cette séparation devrait être renforcée. Cette attitude est révélatrice d'une réticence, sinon d'un refus, de tirer toutes les conséquences de la création de l'Agence : associer le moins possible cette dernière au suivi des décisions, à la méthodologie des contrôles, aux plans de surveillance, aux questions technologiques qui apparaissent en aval. La D.G.A.L. transmet maintenant - enfin - les plans de surveillance, mais cela ne représente qu'une faible partie des éléments de l'ensemble de l'action de contrôle. La tentation pour l'Agence est alors réelle, devant ce qui peut se caractériser comme un mauvais vouloir, de tenter de s'approprier ce qu'elle appelle « l'évaluation de la gestion du risque ». La sémantique administrative est aussi un outil pour décrypter ces conflits de frontière qui sont d'abord des conflits de pouvoir.
Le ministère de l'agriculture n'est pas le seul à manifester ces réticences, mais c'est lui qui le fait le plus nettement. La position de la direction générale de la santé est par nature différente de celle des deux autres tutelles ; celle de la D.G.C.C.R.F. illustre au mieux la difficulté, mais aussi la possibilité réelle, d'établir un équilibre dans ces relations de coopération mutuelle entre l'Agence et les services des trois ministères de tutelle.
La D.G.C.C.R.F. envoie systématiquement les programmes, les plans de contrôle et la méthodologie utilisée à l'AFSSA. Ses remarques sont prises en compte et si à ses yeux le fonctionnement de ce système peut faire l'objet de retouches, il n'y a pas lieu de le modifier actuellement.
Par ailleurs, le système d'échange d'informations au niveau européen par le réseau Rapex, dont la direction générale « Santé des consommateurs » assure la régulation depuis Bruxelles, constitue un élément essentiel et des améliorations pourraient même être obtenues. Certains Etats-membres communiquent des données excessivement abondantes, d'autres à l'inverse des données lacunaires ou équivoques (cas de la dioxine dans les élevages de poulets en Belgique). La Direction générale de l'alimentation, comme la D.G.C.C.R.F. participent à ce réseau.
Les moyens de coopération pour une meilleure efficacité existent, mais là aussi plus qu'une science, la coopération dans la gestion du risque est un art dont la valeur réside essentiellement dans l'application ; si l'AFSSA a rendu immédiatement publique une appréciation en forme d'avis sur les premiers plans de contrôle de la viande bovine sans attendre la réponse des tutelles, il n'est pas étonnant qu'ensuite des réticences regrettables se fassent jour. Dans l'autre sens, il est à noter que deux ans avaient été nécessaires pour que l'AFSSA obtienne communication des modes opératoires d'enlèvement des produits dangereux dans les carcasses de bovins. S'il apparaît assez nettement que tous les torts ne sont pas du même côté, il est à craindre que la tentation du recours à l'argutie soit également bien répartie.
Le genre de blocages que l'on vient d'évoquer appelle des remises en cause de la séparation évaluation/gestion du risque du côté de l'AFSSA. On pointe alors l'argument selon lequel le format des données collationnées diffère selon la perspective d'utilisation : la logique du contrôle n'est pas forcément celle de l'évaluation du risque. La portée de cet argument ne paraît importante que si l'on se place dans une logique de conflit entre unités administratives. Des mécanismes d'échange de données fondés sur des moyens de traitement dans une perspective de coopération mutuelle ne constituent pas un objectif inaccessible ...
Par ailleurs, le contrôle et la répression ne peuvent être séparés et s'inscrivent nécessairement dans le suivi de la gestion du risque. Sauf à placer l'ensemble des unités administratives, laboratoires compris, dans le périmètre de l'AFSSA et à faire disparaître en conséquence la Direction générale de l'alimentation et la D.G.C.C.R.F., il n'apparaît pas aujourd'hui nécessaire de revenir sur l'équilibre établi par la loi de 1998. En revanche, les déviations ou les non-applications doivent être redressées. Des propositions seront faites en ce sens. Une synergie peut se réaliser entre les deux catégories d'acteurs : l'AFSSA d'un côté, les trois administrations chargées de la gestion, de l'autre. Une coopération structurée oblige chacun à préciser ses intentions, sa méthodologie et aussi les limites de sa capacité d'appréciation. A cet égard, un des éléments de la clarification à opérer peut être fourni par le contrat d'objectifs et de moyens (COM) dont l'AFSSA a demandé avec insistance la réalisation depuis longtemps et dont la conclusion a été reportée à plusieurs reprises pour des raisons obscures qui sont plutôt des prétextes. Un outil administratif de ce type n'est sans doute pas la panacée, mais en permettant de préciser les moyens, les exigences et les obligations de chacun, il contribue nécessairement à l'efficacité de l'ensemble des actions dans le cadre général de l'objectif commun qu'est la sécurité sanitaire alimentaire.
2.3. Les saisines : un mécanisme perfectible
Le mécanisme de fonctionnement par saisine illustre bien le caractère novateur de la loi du 1 er juillet 1998 par l'autonomie qu'elle confine aux agences sanitaires, notamment à l'AFSSA, et par la procédure formelle qu'elle établit ainsi (cf. supra première partie).
Au-delà de la satisfaction générale constatée sur ce dispositif, satisfaction que nous partageons, des améliorations sensibles paraissent pouvoir être apportées soit pour faire face à quelques imperfections, soit pour prévenir des risques de dérives actuellement discrets, mais qu'il convient précisément de repérer. Enfin, quelques interrogations demeurent sur certains points où des propositions concrètes peuvent être faites ou envisagées.
2.3.1. Des perfectionnements possibles
Rapidement après sa mise en place en 1999, l'AFSSA a su atteindre un rythme d'activité élevé et faire face aux nombreuses demandes qui lui étaient adressées et ce, au-delà des dossiers les plus brûlants et les plus médiatisés dans des délais très convenables. Pour que la procédure de saisine et de communication des avis garde toute sa valeur et son caractère opérationnel, plusieurs pistes de perfectionnement sont à envisager, notamment d'un point de vue quantitatif.
2.3.1.1. Un volume à maîtriser
Depuis 2000, le nombre de saisines est resté stable à un niveau que l'on peut qualifier d'élevé : entre 330 et 360 par an, mais il s'est élevé à 399 en 2003. Plus de la moitié portent sur des demandes d'autorisation (évaluation des risques dans le cadre d'une mise sur le marché), cette catégorie étant en baisse par rapport au début du fonctionnement de l'Agence ; une proportion stable de 25 % s'observe pour les saisines sur les textes réglementaires et assimilés ; on note une augmentation importante en 2001 des demandes d'évaluation dans des situations de contamination avérée ou potentielle. Le pourcentage de saisines à traiter en situation d'urgence reste constant.
La répartition selon les domaines visés par les saisines en moyenne au cours des trois dernières années est la suivante :
- 25 % sont relatives au domaine de l'eau
- 18 % celui de la nutrition
- 15 % celui de l'alimentation animale
- 10 % celui des ESST (encéphalopathie spongiforme subaigüe transmissible), le reste dans les différents autres domaines de compétence de l'Agence.
L'accent mis sur nombre de saisines par l'Agence elle-même, mais aussi la tendance générale à évaluer principalement une entité administrative à partir de tels critères quantitatifs comporte des risques. Tout en soulignant la nécessité de disposer d'éléments d'information concrets comme celui-ci, il convient d'en mesurer les limites et en tout cas d'introduire les nuances qui permettent d'affiner le jugement et d'abord d'éviter les erreurs.
L'énergie mobilisée au sein de l'Agence elle-même, y compris dans les comités d'experts spécialisés est évidemment très variable selon la nature du questionnement et le domaine concerné. Si l'on souhaite éviter que cette énergie soit diluée ou mise en oeuvre dans des conditions qui limitent l'efficacité de l'action de l'Agence, il convient de concentrer les saisines de l'Agence sur l'essentiel. Deux pistes pour ce cadrage quantitatif sont à envisager.
2.3.1.2. L'examen excessif des textes réglementaires
La consultation sur les projets de textes réglementaires
25 % des saisines sont relatives à l'analyse des projets de textes réglementaires y compris européens dans tous les champs de compétence qui relèvent de l'AFSSA, dont l'étendue, la variété et la technicité n'ont pas besoin d'être davantage explicitées. Le pouvoir qui a été donné à l'Agence se justifie aisément par la volonté de s'assurer le concours permanent d'une compétence reconnue qui a en charge le suivi des dossiers scientifiques visés par ces textes. La détermination de cette compétence d'une manière très explicite (art. L 1323-2, 2è alinéa du CSP peut aussi s'apprécier comme la volonté d'asseoir clairement le rôle de l'AFSSA dans l'élaboration des normes en supprimant dès l'origine toute occasion de querelle de frontières avec les différentes administrations de tutelle et autres.
La pratique en usage depuis maintenant plus de quatre ans montre que cette tâche tend à devenir excessive par l'énergie quelle exige et pas toujours justifiée quant à l'intérêt d'une intervention qui a pris un caractère systématique. C'est sans doute là que réside une partie de la difficulté, mais aussi l'élément de solution. Le texte précité indique que « l'Agence fournit au Gouvernement l'expertise et l'appui scientifique et technique qui lui sont nécessaires ... » ; est-il nécessaire dans tous les cas où l'Agence est saisie ? On peut en douter. Une sélection pourrait être effectuée afin de limiter sensiblement le nombre et éventuellement l'ampleur de ces saisines qui, à tort, sont devenues systématiques.
Dans certains domaines bien déterminés, la saisine de principe de l'Agence doit entièrement être maintenue. C'est le cas évidemment pour tout ce qui est relatif au médicament vétérinaire où l'AFSSA est détentrice de la décision (AMM), du pouvoir de police et de contrôle. La même démarche doit être retenue pour tous les projets de textes et de décisions dans le domaine européen et international : la nécessaire et souvent difficile coordination interministérielle sur des sujets qui sont scientifiquement complexes et commercialement disputés ne pourrait se passer de la compétence de l'AFSSA. C'est parmi ce qui reste qu'un allègement pourrait être opéré.
2.3.1.3. Des saisines contestables
-- A travers des consultations sur des projets de textes sont posées des questions dont la réponse est cadrée par les valeurs limites préexistantes (denrées alimentaires d'origine animale contaminées par des résidus de pesticides). D'autres cas relèvent de la gestion courante du risque et n'appellent pas autre chose que la confirmation de l'application des dispositions déjà applicables : c'est particulièrement le cas dans le domaine de la santé animale ; des textes relatifs au transport des animaux relèvent de la même analyse ; l'existence au sein de l'AFSSA d'un « directeur de la santé animale » disposant d'un statut particulier par rapport au ministère de l'Agriculture est peut-être l'explication de ce type de saisine.
-- Il arrive que l'intitulé de la saisine montre qu'une telle demande ne paraît pas relever de ce traitement administratif : ainsi en 2003, la saisine 124 : « demande d'avis relatif à trois projets d'arrêtés concernant la gestion des sous-produits animaux non destinés à la consommation humaine » exposée par la DGAL.
-- Un tout autre problème est posé par celui des saisines visant à faire valider des justificatifs d'allégation nutritionnelle ou de santé proprement dite. C'est là un phénomène récent, en rapide développement que l'on abordera par ailleurs. Mais d'ores et déjà il apparaît que ces demandes dont l'admissibilité même pose problème, et qui émanent le plus souvent « d'entreprises fugitives », devraient être découragées ; et en tout cas elles devraient faire l'objet d'une « procédure accélérée » qui pourrait précisément être instituée à cette fin.
-- Certaines saisines relatives à des renouvellements d'agrément de matériaux dans le traitement des eaux peuvent relever d'une autre procédure administrative, car on est ici dans la gestion plus que l'évaluation.
-- Au total, en regroupant les différents éléments qui viennent d'être identifiés, il apparaît que le nombre de saisines pourrait être diminué de 15 % à 18 % sur la totalité (399) de celles enregistrées en 2003, qui est d'ailleurs en augmentation sensible sur les années précédentes.
2.3.1.4. Les moyens d'une maîtrise du volume des saisines
L'identification des imperfections donne d'elle-même les pistes d'améliorations possibles.
L'automaticité de saisine sur les textes, notamment ceux qui relèvent d'une routine d'application et non d'une évaluation scientifique ou technologique nouvelle, peut être évitée. Les administrations de tutelle ont évidemment leur part de responsabilité dans cette réorientation. Il en est de même pour des « saisines techniques » dont la recevabilité au titre de l'évaluation n'est pas évidente.
Dans les autres cas, c'est sans doute par un rôle accru, voire nouveau, du Conseil scientifique que des progrès substantiels dans ce domaine pourraient être obtenus. Une nouvelle tâche pourrait lui être donnée dans le cadre de l'art. R 794-22 du Code (formation restreinte) lui permettant de jouer un rôle de tamisage dans le flux des saisines. Les comités spécialisés d'experts pourraient être aussi amenés à intervenir selon une procédure simplifiée par rapport à la procédure normale, d'autant que le secrétariat scientifique de chacun de ces comités, dont le fonctionnement est jugé tout à fait satisfaisant, pourrait y contribuer.
La maîtrise quantitative des saisines contribuera à améliorer la qualité d'un dispositif qui répond sans aucun doute aux objectifs qui lui ont été fixés, mais qui, si l'on n'y prenait garde, pourrait connaître des dérives que l'augmentation excessive du nombre de saisines pourrait notamment susciter.
2.3.2. Des risques de dérives
2.3.2.1. La tendance naturelle de notre époque à considérer le risque zéro comme un objectif accessible, donc souhaitable 25 ( * ) , peut très naturellement amener à multiplier les précautions, les vérifications répétées dont l'intitulé de certaines saisines de l'AFSSA témoigne d'ailleurs clairement. Il n'y a donc rien d'étonnant à ce que les administrations de tutelle contribuent à une augmentation sensible et pas toujours justifiée des saisines. Ce mouvement peut même avoir une origine directement ministérielle : la tentation de « se couvrir » reste forte dès lors que l'on sent l'opinion d'une extrême sensibilité à un problème. En outre, le mécanisme fonctionnant bien, il n'y a pas vraiment de régulation naturelle dans le sens de la maîtrise du flux qui puisse s'établir. La croissance constatée en 2003 peut procéder de ce phénomène, mais surtout elle pourrait être plus sensible dans l'avenir.
Par ailleurs, d'autres facteurs qui vont dans le même sens sont identifiés. L'AFSSA elle-même pointe la croissance que pourrait entraîner la réalisation effective de la séparation entre l'évaluation et la gestion du risque dans les domaines des produits biocides et phytosanitaires. C'est là un problème essentiel que l'on aborde plus loin dans le présent rapport (« des lacunes à combler ») mais dont on peut dès maintenant prévoir les implications éventuelles pour l'AFSSA.
2.3.2.2. Les organisations de consommateurs ont depuis décembre 2000 la possibilité de saisir l'AFSSA ; elles en ont fait un usage très limité (moins d'une dizaine par an). On peut penser que l'on observera ici une croissance, mais on ne dispose pas d'indication précise pour le moment.
2.3.2.3. Les industriels ne disposent pas du droit de saisine. L'ANIA (Association Nationale des Industries Alimentaires) et, plusieurs autres acteurs, souhaitent que ce droit leur soit reconnu. Il s'agirait d'un élément pouvant contribuer à la croissance du nombre des saisines, mais il conviendrait, le cas échéant, d'éviter une dérive dans cette perspective (cf. infra).
2.3.2.4. Enfin, si les auto-saisines de l'AFSSA ne sont pas statistiquement considérables (20 sur 347 en 2001), leur évolution et surtout la charge qu'elles représentent (cf. supra) sont de nature à mobiliser une part importante de l'énergie de l'Agence. Or l'autosaisine n'étant pas encadrée, elle peut apparaître et se développer dans des domaines et des modalités qui sont discutables. Les efforts qui y sont consacrés peuvent l'être au détriment de travaux plus nécessaires ou plus légitimes. Les évolutions inquiétantes caractérisées par l'augmentation de la prévalence de l'obésité, ont conduit à mettre l'accent sur les actions dans le domaine nutritionnel. Ce souci justifié ne saurait entraîner une déformation de l'outil qu'est l'Agence modifiant ainsi la répartition des moyens qui sont mis à sa disposition par rapport à l'ensemble de ses objectifs et de ses obligations.
2.3.3. Des interrogations
2.3.3.1. Sur le droit de saisine
La question du droit de saisine de l'AFSSA par les industriels (producteurs, distributeurs) est régulièrement soulevée par tous les acteurs : les intéressés bien sûr, mais aussi les administrations, les experts et les autres parties prenantes. L'ANIA (Association Nationale des Industries Alimentaires) avait demandé que cette faculté leur soit reconnue et en janvier 2003, le CNA (Conseil National de l'Alimentation) a émis un voeu en ce sens.
Dans la mesure où les organisations de consommateurs en bénéficient, on voit mal comment ce droit pourrait par principe être refusé à bien des acteurs essentiels du domaine, sauf à se fonder sur une idéologie d'exclusion des intérêts privés alors qu'il s'agit d'un droit de saisine et non d'une quelconque participation à la décision ou à la gestion.
Parmi les arguments invoqués, et en dehors des industriels eux-mêmes, on a souvent évoqué le fait que la nouvelle architecture de la sécurité sanitaire des aliments avait marginalisé les industriels et les experts du secteur privé. Au-delà des questions purement scientifiques, les applications technologiques semblent, de ce fait, avoir été moins prises en compte. Cette évolution pourrait avoir des conséquences sur la valeur de l'expertise sur l'ensemble des process et de ce fait dans l'appréciation des risques eux-mêmes. Le droit de saisine, logique au demeurant, serait donc un moyen de limiter les effets de cette séparation dommageable. Dans ses propositions, le CNA prend en compte lui-même les impératifs et les précautions qu'implique cette réforme :
Sur le champ du droit de saisine
La possibilité de saisine de l'AFSSA devrait porter sur l'évaluation des risques suspectables compte tenu des éléments dont disposent une ou plusieurs entreprises d'un même secteur. Les exploitants du secteur alimentaire devraient, dans ce cadre, pouvoir obtenir de l'AFSSA un avis sur les risques nutritionnels ou sanitaires qu'ils suspecteraient concernant les aliments destinés à l'homme ou aux animaux, les procédés et conditions de production, transformation, conservation, transport, stockage et distribution des denrées alimentaires, en informant simultanément les autorités sanitaires compétentes.
Cette saisine devrait dans tous les cas s'inscrire dans une perspective d'intérêt général de santé publique et non d'intérêt particulier.
Ce droit de saisine ne devrait pas pouvoir s'exercer dans les domaines où les textes en vigueur prévoient une procédure de dépôt de dossier ou de demande d'autorisation auprès d'une administration.
Sur l'exercice du droit de saisine
Pour éviter tout risque de saturation de l'Agence ou des saisines qui tendraient à détourner l'Agence de son rôle institutionnel et à lui faire jouer un rôle de conseil, le CNA recommande que cette possibilité soit ouverte aux seules organisations professionnelles ou interprofessionnelles.
S'agissant d'une simple faculté, il serait loisible à une organisation professionnelle ou interprofessionnelle, si elle estime que de telles saisines ne relèvent pas de son rôle ou qu'elle n'est pas en mesure de les prendre en charge, d'informer ses mandants de cette position de principe.
Ce n'est qu'au terme d'une période expérimentale, par exemple de deux ans, que la possibilité d'ouvrir le droit de saisine aux exploitants eux-mêmes pourrait être examinée, tout en tenant compte des mêmes préoccupations.
Ce texte prévoit la possibilité pour l'Agence de juger de la recevabilité de la saisine et, de ce fait, de la rejeter :
« Si la demande n'entrait pas dans le domaine de compétences de l'Agence, si elle ne relevait pas de l'évaluation du risque, si elle était insuffisamment motivée et/ou renseignée ou si elle ne relevait pas de l'intérêt général de santé publique, mais d'un intérêt strictement privé, il informerait le demandeur que sa demande n'est pas susceptible d'être examinée ».
L'ouverture d'un tel droit pose, en effet, quelques questions difficiles que le CNA a bien identifiées, notamment le risque d'instrumentalisation ou de saturation de l'Agence. Les précautions envisagées paraissent de nature à y répondre. Il reste que le fait de faire filtrer le droit de saisine par le canal des organisations professionnelles peut soulever des difficultés en terme de rapports de force d'une part, et au regard du secret industriel de l'autre. Ces réserves ne constituent pas une raison suffisante pour ne pas réaliser une réforme dont la quasi totalité des acteurs s'accordent à noter le bien fondé.
2.3.3.2. Sur le champ des saisines
On a vu que l'Agence elle-même notait clairement parmi les évolutions envisageables l'évaluation dans le domaine des produits phytosanitaires et celui des biocides. Sans nous prononcer actuellement sur le second, les questions soulevées par le premier ne peuvent qu'avoir des conséquences sur la tâche de l'Agence dans un domaine où elle est expressément chargée par les textes de la sécurité sanitaire.
L'aménagement des compétences de l'AFSSA et de l'AESA
La création effective de l'AESA, il y a près de deux ans, implique la détermination des champs de compétences avec les agences nationales. On traitera spécifiquement ce problème plus loin, mais il convient de noter dès maintenant que de réels problèmes se posent déjà et qu'ils ont des conséquences sur la conception des saisines donc des avis de l'AFSSA. Quant au nombre, il est sans doute beaucoup trop tôt pour délimiter les effets d'un changement dans l'ordonnancement institutionnel encore difficile à cerner.
2.4. L'expertise : des retouches nécessaires
L'expertise est l'élément essentiel de l'évaluation qui elle-même est le coeur de métier de l'AFSSA. Elle doit répondre aux exigences croissantes d'une population de plus en plus informée. La mise en place de l'Agence ayant été abordées précédemment, on se limitera ici aux appréciations relatives aux années récentes et aux questions plus durables, sachant d'ailleurs que celles-ci relèvent autant de mise en oeuvre de principes et d'organisation interne que de dispositions législatives. On est tout au plus dans le domaine réglementaire. Mais de toute façon, la réalité de terrain, celle du fonctionnement de l'expertise, présente davantage d'intérêt qu'une approche étroitement juridique.
Une précision liminaire doit enfin être donnée : il ne revient évidemment pas au rapporteur de l'Office de se livrer à une appréciation de la valeur scientifique des expertises réalisées ; il n'en a naturellement pas les moyens ; une telle appréciation ne saurait se faire à partir d'exemples limités. Les corps d'inspection (IGAS et IGF) eux-mêmes, saisis d'une demande d'audit sur l'AFSSAPS en 2002 ont de la même façon exclu toute capacité à procéder à une telle évaluation dans le domaine des produits de santé. En outre, l'évaluation dont nous sommes chargés ici n'est pas un audit.
Après ces remarques et quelques interrogations sur la situation des experts eux-mêmes, on abordera le fonctionnement des comités d'experts et leur place dans l'ensemble du dispositif d'évaluation du risque.
2.4.1. La question des experts
La qualité et l'indépendance des experts constituent des éléments acquis dont on a rappelé la réalité dans la première partie du présent rapport ; on abordera essentiellement ici les remarques que certains aspects de leur situation soulèvent.
2.4.1.1. La situation
La situation naturelle et les conditions de travail des experts ont été sensiblement améliorées par rapport à la période antérieure à la création de l'AFSSA. Ce progrès sensible n'est contesté par personne. Il répondait d'ailleurs à un besoin réel. Ceux qui ont connu l'époque de la section « alimentation nutrition » du conseil Supérieur d'hygiène publique de France en attestent. Il est vrai qu'on y a mis quelques moyens.
Cela posé, la rémunération ne semble pas être l'élément le plus susceptible d'attirer un grand nombre d'experts dans les comités spécialisés et l'AFSSA elle-même indique que dans l'analyse de coût des avis qu'elle a réalisée, les vacations des experts ne représentent que 6 % (et les frais de transport et de mission afférents 7 %).
La situation ne semble pas actuellement inquiétante ; toutefois, lors du premier renouvellement des comités d'experts (250 personnes au total) intervenu en 2003, on a enregistré un nombre de candidats en nette diminution par rapport au recrutement précédent.
La charge de travail peut être très lourde dans certains comités. Un président de comité d'experts indique, par exemple, qu'il a eu à rendre 120 avis en deux ans et demi. Il n'est pas rare que des experts estiment que leur activité pour l'AFSSA « relève du bénévolat », compte tenu de ces éléments.
Enfin, la reconnaissance que manifeste l'Agence elle-même aux experts ne semble pas être à la mesure des efforts importants qu'ils lui consacrent ; le rapport 2002-2003 de l'AFSSA en donne un signal très clair : l'activité des comités d'experts est évoquée d'une manière particulièrement elliptique, à travers le soutien que leur apportent les scientifiques de l'AFSSA et plus nettement par le faible volume de leurs vacations.
2.4.1.2. La carrière
La « motivation de carrière » n'existe pas réellement pour ce type d'activité et ce, d'une manière nettement plus marquée que pour le médicament. En effet, dans les disciplines scientifiques intervenant dans le domaine alimentaire, le travail d'expertise n'est absolument pas pris en compte à sa juste valeur ; il peut même être méprisé par rapport aux activités de recherche qui, d'un point de vue académique, n'ont évidemment pas le même « statut ». Certains scientifiques se voient ainsi reprocher de « faire trop d'expertise ». Le risque est aussi que progressivement on ne recrute pas les experts parmi les meilleurs. Enfin, un expert lui-même observe « cette activité constitue une responsabilité à une époque où l'on ne recherche pas tellement cela ».
Cette insuffisance de reconnaissance ne touche naturellement pas seulement l'AFSSA, ce qui n'est pas rassurant sur un plan général et pour l'avenir.
Il y a lieu de noter d'ailleurs que la France, malgré sa position dans le domaine agricole et alimentaire, n'est pas le pays européen qui fournit proportionnellement le plus d'experts : le Danemark, la Grande-Bretagne et les Pays-Bas, la dépassent en effet. Cette constatation devrait faire d'autant plus réfléchir pour que cette tendance ne n'accentue pas, compte tenu en outre du développement prévisible de l'expertise au niveau européen (AESA) et mondial (JECFA entre autres). Le Parlement n'a pas ici de pouvoir de décision, mais il a le devoir d'attirer l'attention sur la nécessité d'agir à différents niveaux et vis-à-vis des différents acteurs (administrations dont l'Agence, monde universitaire). Ces insuffisances de considération et dans certains cas d'effectifs ont une traduction plus aiguë dans certaines spécialités, notamment la toxicologie.
2.4.1.3. Les experts non académiques
L'absence d'experts « non académiques » travaillant dans le secteur industriel pose un problème réel. La clarification de la situation des experts par rapport aux intérêts industriels en est directement à l'origine. L'effectivité du mécanisme des « déclarations d'intérêt », la volonté de mettre un terme à des situations abusives dont l'existence était reconnue avant 1998, ont entraîné une coupure particulièrement marquée entre l'industrie et l'expertise dans le cadre de l'évaluation du risque. Ainsi, la « commission de technologie alimentaire de l'ancien CSHPF a été supprimée avec la création de l'AFSSA. Dans certains domaines comme celui des emballages ou des arômes, il est difficile de se passer de la participation d'experts oeuvrant dans la production. On est certainement allé trop loin en refusant de considérer les exigences des réalités techniques. En outre, cela limite considérablement les effectifs d'experts, aggravant encore le problème de la limitation du « vivier de recrutement » que l'on vient d'analyser. Les précautions à prendre exigent du soin dans l'édiction des règles et le recours des experts exige des précautions ; cela n'est pas une raison pour continuer à feindre d'ignorer un problème réel qui, avec le développement de technologies de plus en plus élaborées et le mouvement de mondialisation des échanges dans le domaine alimentaire, ne pourra que s'aggraver.
2.4.2. Le fonctionnement des comités d'experts
Leur mise en place réalisée, les comités d'experts ne connaissent pas de difficultés marquantes dans leur fonctionnement. Mais les experts eux-mêmes posent d'une manière récurrente des questions qu'il convient d'entendre.
L'ampleur du périmètre de compétences des comités d'experts spécialisés peut être l'objet d'une interrogation : comités de spécialistes ou comités plus « pointus » de spécialistes.
2.4.2.1. Des appréciations diverses
Le fonctionnement lui-même est apprécié de manière diverse. L'inégalité de situation des différents comités l'implique sans doute, mais quelques questionnements sont communs : variété du traitement des saisines, place de l'expertise interne, articulation entre l'avis du comité et celui de l'AFSSA.
Le document d'évaluation de l'AFSSA réalisé à la demande de celle-ci par le Centre de sociologie des Organisations (CNRS-FNSP) 26 ( * ) recèle sur ce point des informations précises qui semblent refléter la situation ressentie par les présidents de comités d'experts. Inséré dans un paragraphe « clarifier certaines étapes du processus d'expertise » ce développement dont on présente ci-après l'essentiel du texte est intitulé « un besoin de clarification du mode de traitement des saisines » :
« Un besoin de clarification du mode de traitement des saisines
Le choix des modalités de traitement des saisines n'est pas très bien compris, aussi bien par le personnel de l'Agence que par les présidents de CES. Le partage des saisines entre travail au sein de la DERNS et travaux en CES n'obéit pas à des procédures clairement établies et énoncées et le choix de l'expertise interne peut alors être mal vécu par les experts des CES ou les secrétaires scientifiques.
En plus de la charge de travail conséquente et d'une situation quasi-bénévole, les experts ont la sensation que le secrétariat scientifique ne fait pas toujours son travail de tri des saisines. Ainsi, ils traitent parfois malgré leur volonté des dossiers qu'ils estiment être non pertinents :
« Le seul problème est l'harmonisation des saisines, il faudrait parfois faire le tri des saisines ; le CES étant très chargé, il ne faudrait pas être sollicité par les mêmes questions trop souvent, ou des questions qui paraissent superflues ». (président de CES).
Certains présidents peuvent ressentir la frustration de ne pas être toujours associés à l'étape du choix de traiter ou non la saisine en CES. Cette étape serait nécessaire pour permettre au président de choisir des questions motivantes pour ses experts. Le flou autour de la procédure alimente les interprétations et le sentiment de non-reconnaissance du travail des experts :
« S'il existait une vraie démarche assurance qualité, le secrétaire scientifique avec les présidents de comité devraient faire une réunion de saisine à l'arrivée pour savoir qu'est-ce qu'on fait. Actuellement les présidents ne sont pas mis à contribution. Je cherche à ce que les experts soient motivés. Alors si on leur donne des sous-saisines, cela ne vaut pas la peine, ils ne viendront plus. Il faut qu'ils aient quelque chose à se mettre sous la dent, qu'ils sentent que leur expertise est mise à contribution. Pour être motivés, il faut qu'ils se sentent indispensables ». (président de CES).
« Il y a aussi un problème de nature des saisines, et de tri des saisines selon leur importance et dans la manière de les traiter. Des petites saisines sans importance dont je pense qu'elles ne devraient pas être traitées en comité passent dans le CES. Et des saisines importantes qui nous auraient intéressées sont traitées totalement par l'agence, sans référence aux CES ». (président de CES).
(...)
Les représentants des administrations de tutelle qui ont été interviewés 27 ( * ) , eux aussi, acceptent mal que l'expertise soit réalisée en interne, sans doute parce qu'elles ont alors moins de visibilité sur le travail effectué, alors que lorsque la saisine est traitée par le CES, du fait de leur présence en comité, elles peuvent suivre le traitement qui en est fait » :
Ces témoignages confirment des remarques déjà faites par ailleurs, mais d'abord elles soulignent effectivement un réel besoin de clarification. La qualité des secrétariats scientifiques assurés par la DERNS ne justifie pas que ses membres au sein des comités exercent une prérogative qui n'est pas forcément la leur, sans que les lignes de partage aient été clairement fixées à l'avance sur ce que traite le comité lui-même directement. Le principe d'une partie d'expertise interne n'est évidemment pas contestable, mais des règles claires doivent être posées, et notamment vis-à-vis des responsables de l'expertise externe que sont les présidents de comité et les rapporteurs.
Par ailleurs, l'avis de l'AFSSA est celui signé par le directeur général de l'Agence. Il peut comporter des différences avec celui rédigé par le comité d'experts spécialisés. Ces modifications éventuelles font généralement l'objet de discussions entre le président du comité et le directeur général, mais cela n'est pas toujours le cas et quelques situations conflictuelles se sont ainsi produites. Il arrive aussi - on l'a observé récemment sur des questions sensibles - que l'avis de l'AFSSA cite les termes de l'avis du comité d'experts, en cultivant l'ambiguïté afin d'imposer un point de vue qui n'est pas strictement celui des scientifiques
2.4.2.2. Les administrations dans les comités d'experts
La présence de représentants de la DGCCRF, de la DGAL et de la DGS dans les comités est assez régulièrement un sujet de discussion convenu, notamment entre non praticiens de ces procédures. Il n'est pas douteux que dans le passé (avant 1998), certaines administrations ont eu tendance, à peser sur l'expertise ou à tenter de le faire. Mais la loi de 1998 a précisément eu pour objectif de mettre fin à ces pratiques et en a donné les moyens. L'objectif a globalement été atteint et les praticiens, tant les experts que la DERNS elle-même, estiment que l'absence des administrations de tutelle dans les comités d'experts serait particulièrement dommageable au fonctionnement efficace de ceux-ci : lacune d'informations, impossibilité de remise en perspective, faisabilité des choix etc ... Peu à peu un modus vivendi s'est établi et les tutelles elles-mêmes se concertent beaucoup mieux. En cas de risque de débordement, le président de chaque comité a tous les moyens de rappeler chacun aux limites de son rôle.
On voit mal comment l'ensemble du mécanisme pourrait fonctionner sans la présence des représentants des administrations. Toute modification substantielle de ce fonctionnement efficace et équilibré compromettrait sérieusement un résultat très positif.
2.5. Une lacune à combler et des faiblesses à redresser
Au-delà des aménagements des procédures ou des pratiques que le comportement des différents acteurs a révélé nécessaires, le volume des saisines, l'exercice de l'auto-saisine ou la communication de l'Agence notamment, il est des secteurs dans lesquels on a à faire à des problèmes d'une autre ampleur.
Dans un cas, il s'agit d'une véritable lacune : les produits phytosanitaires ; dans d'autres, il s'agit plutôt de faiblesses. En témoignent avec les difficultés que pose le système complexe du traitement des dossiers d'OGM par de trop nombreux intervenants ; c'est aussi le problème posé par les allégations santé.
2.5.1. Le cas des produits phytosanitaires
La loi du 1 er juillet 1998, par l'art. L 1323-1 du code de la santé publique a clairement défini la compétence de l'AFSSA dans sa mission fondamentale « d'assurer la sécurité sanitaire dans le domaine de l'alimentation depuis la production des matières premières jusqu'à la distribution et au consommateur final ». Le texte précise ensuite :
« Elle évalue les risques sanitaires et nutritionnels que peuvent présenter les aliments destinés à l'homme ou aux animaux, y compris ceux pouvant provenir des eaux destinées à la consommation humaine, des procédés et conditions de production, transformation, conservation, transport, stockage et distribution des denrées alimentaires, ainsi que des maladies ou infections animales, de l'utilisation des denrées destinées à l'alimentation animale, des produits phytosanitaires, des médicaments vétérinaires, notamment les préparations extemporanées et les aliments médicamenteux, des produits antiparasitaires à usage agricole et assimilés, des matières fertilisantes et supports de culture, ainsi que des conditionnements et matériaux destinés à se trouver en contact avec les produits susmentionnés. De même, elle participe à la mission de défense nationale dans le domaine alimentaire ».
La lettre et l'esprit de la loi sont clairement exprimés ; les auteurs de la proposition de loi sénatoriale (MM. Charles Descours et Claude Huriet), et les différents intervenants au débat parlementaire ont toujours envisagé que ce domaine essentiel que sont les produits phytosanitaires, les produits antiparasitaires à usage agricole et assimilés, soient à l'intérieur du périmètre de la loi et de la responsabilité de l'Agence.
La réalité est éloignée de cette architecture générale voulue par le législateur. Il y a là une faiblesse majeure dans la mise en oeuvre de la loi, d'autant qu'il s'agit d'un sujet particulièrement transversal et que la participation des acteurs autres que le ministère de l'agriculture n'est pas mieux assurée que celle de l'AFSSA.
Des précisions en forme de rappel sont ici nécessaires, la subtilité des questions scientifiques s'ajoutant à la complexité de l'ordonnancement administratif et à l'opacité du fonctionnement de l'ensemble. Il convient donc de décrire le dispositif actuellement en fonction.
Les instances d'expertise intervenant pour l'évaluation des risques relèvent du ministère de l'agriculture, de l'alimentation, de la pêche et des affaires rurales. Malgré la réforme que comporte la loi de 1998 (cf. texte supra), l'essentiel n'a pas changé, c'est-à-dire que la commission d'étude de la toxicité des produits phyto-sanitaires reste sous la tutelle du ministère de l'agriculture : il n'y a donc pas là séparation entre l'évaluation et la gestion du risque.
Cette commission d'étude de la toxicité des produits antiparasitaires à usage agricole et des produits assimilés (Comtox) est chargée de l'évaluation des risques pour la santé et l'environnement liés aux intrants en agriculture (produits phytopharmaceutiques - encore appelés pesticides - mais aussi matières fertilisantes soumises à autorisation). Elle comprend 35 experts et s'appuie sur son réseau de 50 experts associés ; des améliorations dans le sens d'une indépendance plus tangible ont été apportées avec l'exclusion des représentants des professionnels, des associations de consommateurs et de protection de l'environnement d'une part, et l'introduction d'un appel public à candidatures pour les experts d'autre part.
Le comité d'homologation, pour les mêmes produits, est compétent pour l'autorisation de mise sur le marché.
La Comtox s'appuie sur le travail d'une structure scientifique et technique (SSM) créée conjointement par l'INRA (Institut National de Recherche Agronomique) et le ministère de l'agriculture. La première partie de l'évaluation porte sur les préparations commerciales contenant une ou plusieurs substances actives.
Cette structure scientifique mixte, est le résultat d'une demande de l'Union européenne dans le cadre de la directive 91/414. Cette création a permis de faire passer de 3 à 15 le nombre de personnes en charge de cette phase d'évaluation, lors de la mise en place en 1997. C'est d'autant plus appréciable que la Comtox elle-même a en charge l'examen de 1200 dossiers par an environ, ce qui, même si l'on tient compte des « produits bis » ne manque pas d'étonner des scientifiques chargés de tâches d'évaluation des risques dans d'autres domaines.
La révision des produits phytosanitaires dans le cadre de la directive 91/414 constitue une tâche importante pour la SSM et donc pour la Comtox ; la structure compétente de l'Etat-membre pour un dossier communique son évaluation à l'Agence européenne de sécurité alimentaire (AESA) qui fait circuler la proposition de décision parmi les structures des Etats-membres n'ayant pas participé à l'évaluation, puis vers la D.G. Sanco (direction générale santé des consommateurs) avant la prise de décision par les représentants des Etats-membres au sein du comité permanent de la chaîne alimentaire et de la sécurité animale (section produits phytopharmaceutiques).
Les exigences au niveau européen contribuent ainsi à cet accroissement de la charge de travail et dans des conditions peu satisfaisantes puisqu'on tente d'imposer une accélération des procédures. Ainsi, une première liste de 100 produits à réviser a été examinée en 5 ans et l'on se propose d'en faire autant dans un laps de temps plus court. Pour les nouvelles demandes, on se propose de passer de 3 mois à 1 mois. L'exigence n'est plus au niveau de la qualité de la décision, mais de la rapidité d'examen.
Le travail de cette structure scientifique mixte et de la commission d'étude de la toxicité n'est pas contestable ; en revanche, les conditions dans lesquelles s'établit le chaînage entre les expertises et les décisions, ainsi que leur suivi recèlent de graves difficultés dont les développements les plus récents ont pris le tour de mise en accusation de tout un système.
La mise en cause des deux produits phytosanitaires que sont, sous leur nom commercial le Gaucho (principe actif : imidaclopride) et le Régent (principe actif : fipronil), dans les troubles qu'enregistrent les apiculteurs dans leur essaims d'abeilles illustre d'une manière spectaculaire la consistance du problème. Sans traiter au fond une question qui relève de l'expertise scientifique, force est de constater les faiblesses, les insuffisances, et pour tout dire certains dysfonctionnements dont le refus de communication de documents. Dans son livre intitulé « Quand les abeilles meurent, les jours de l'homme sont comptés » paru en février 2003, le député Philippe de Villiers, décrit le développement de ce que l'on peut appeler au moins une série de graves dysfonctionnements qu'il qualifie de « scandale d'Etat ». Sans entrer dans le détail d'un enchaînement précis de faits et de pratiques en effet insolites dans des procédures de ce type, qui ont notamment amené des juges à exiger la communication de dossiers d'expertise dont le ministère de l'agriculture refusait la communication, on peut rappeler quelques faits dans une brève chronologie.
Dès décembre 1997, un rapport concernant le Gaucho présenté à la Commission d'étude de la toxicité s'inquiétait des imprécisions fournies par le producteur Bayer, et d'une durée de persistance du produit dans le sol totalement hors des normes européennes.
En janvier 1999, le ministre de l'agriculture retire provisoirement l'homologation du Gaucho pour le tournesol. Le Conseil d'Etat confirme cette décision en décembre 1999 en rejetant le pourvoi de Bayer (producteur du Gaucho). En octobre 2002, sur la requête d'un syndicat d'apiculteurs (d'avril 2001), le Conseil d'Etat annule la décision par laquelle le ministre de l'agriculture a refusé d'interdire le Gaucho pour le maïs et enjoint au ministre de réexaminer sa position dans un délai de trois mois. Le ministère en fait autorise le 21 janvier 2003 de nouvelles expérimentations dans un communiqué de presse que le Conseil d'Etat analysera comme un refus d'abroger l'AMM pour le Gaucho maïs. En effet, le 31 mars 2004, est rendu un arrêt qui annule ce refus (pour le maïs) dans des termes qui caractérisent bien l'acharnement de l'administration et du ministre, y compris après un premier arrêté.
Extrait de l'arrêt du Conseil d'Etat (séance du 17 mars, lecture du 31 mars 2004)
« Considérant que, par une décision en date du 9 octobre 2002, le Conseil d'Etat statuant au contentieux a annulé le refus du ministre de l'agriculture d'abroger l'autorisation de mise sur le marché du produit dénommé « gaucho » pour les semences de maïs, délivrée pour dix ans le 6 février 1992 et renouvelée pour la même durée le 21 janvier 2002 ; qu'à la suite de cette décision, le ministre de l'agriculture a publié le 21 janvier 2003 un communiqué de presse qui doit s'interpréter comme maintenant son refus d'abroger l'autorisation de mise sur le marché du gaucho pour les semences de maïs, conformément aux avis émis le 18 décembre 2002 et le 20 décembre 2002, respectivement par la commission d'étude de la toxicité des produits antiparasitaires à usage agricole et par le comité d'homologation de ces produits (...).
« Considérant qu'il résulte de tout ce qui précède que l'appréciation à laquelle se sont livrés la commission d'étude de la toxicité et le comité d'homologation et sur la base de laquelle le ministre a pris sa décision sont fondées sur une méthode d'évaluation du risque qui n'est pas conforme à celle qu'exige l'arrêté interministériel précité du 6 septembre 1994 ; que par ailleurs le ministre ne fait pas état de ce que l'autorisation qu'il a refusé de retirer aurait été elle-même fondée sur la méthode légalement exigée ; que par suite la décision attaquée est entachée d'erreur de droit et doit être annulée ;
Sur les conclusions aux fins d'injonction :
« Considérant que la présente décision a nécessairement pour conséquence d'obliger le ministre à statuer à nouveau sur la demande d'abrogation présentée par les organisations requérantes ; que, compte tenu de l'état d'avancement des expertises et notamment du rapport de synthèse établi par le « Comité scientifique et technique de l'étude multifactorielle des troubles des abeilles » au mois de novembre 2003 et produit à l'instance par les requérants, cette décision devra intervenir dans le délai de deux mois ;
(...)
Décide
Article 2 : La décision du ministre de l'agriculture en date du 21 janvier 2003 refusant d'abroger l'autorisation de mise sur le marché du produit dénommé « gaucho » pour les utilisations relatives au maïs est annulée.
Les conditions de l'évaluation à laquelle ont procédé la commission d'étude de la toxicité et le comité d'homologation sont donc très directement mises en cause par l'arrêt ; l'analyse du texte des « considérants » entre dans le détail des critiques méthodologiques et juridiques que les travaux de ces instances et la procédure suivie par le ministre appellent.
La citation des expertises et du rapport de synthèse établi par le « comité scientifique et technique de l'étude multifactorielle sur les troubles des abeilles », montre comment le ministère face à des difficultés et des oppositions qu'il a largement sous-estimées a tenté de résoudre les problèmes par la création de ce comité ad hoc en 2000, à côté des structures existantes. Ce comité a remis son rapport final au ministre en septembre 2003 et il a notamment signalé les risques constatés par l'enrobage « Gaucho » tant pour les semences de maïs que celles de tournesol.
Sur ce sujet, les travaux des laboratoires du CNRS, de l'INRA et de l'AFSSA dont les résultats, déjà diffusés dans le public, avaient servi d'élément d'appréciation dans la première décision d'interdiction du Gaucho sur le maïs le 23 octobre 2002 et ils établissaient clairement la nocivité des deux produits (Gaucho et Régent) pour les abeilles. Il est donc d'autant plus étonnant que soit intervenu en janvier 2002 le renouvellement pour 10 ans de l'homologation du Gaucho, alors même que la procédure d'annulation devant le Conseil d'Etat était en cours ; l'enquête pénale déjà ouverte sur ce produit a d'ailleurs été étendue à ces faits.
L'atmosphère particulièrement lourde dans laquelle ces affaires se sont développées mérite d'être relevée et notamment les comportements de l'administration en cause, le ministère de l'agriculture et plus spécialement la direction générale de l'alimentation. Une proportion importante des chercheurs travaillant sur ces problèmes ont rencontré des difficultés ou ont été l'objet de pressions. Des rapports d'expérimentations des dossiers d'instruction de demandes d'homologation n'ont pu être connus qu'après réquisition des deux juges en charge des enquêtes pénales, particulièrement pour le Régent.
Parallèlement au problème du Gaucho, celui du fipronil, principe actif du Régent, a en outre illustré les dysfonctionnements des procédures au sein de la direction générale de l'alimentation, la faiblesse de la coopération avec les autres entités administratives dans un domaine où elle devrait s'imposer et sur un sujet qui a fini, au-delà des abeilles, par soulever le problème de la dangerosité pour l'homme.
Le Régent TS a bénéficié d'une autorisation provisoire de vente du 1 er décembre 1995 valable quatre ans et renouvelable une fois pour deux ans. Une « autorisation provisoire de vente » a été accordée le 11 mars 2003, à la société Cropscience, bien après l'expiration de celle de 1995 (quatre ans) et selon une procédure qui ne paraît pas conforme à celle qui est applicable à ce produit, compte tenu de certaines de ses caractéristiques. Il a en effet à la même date été classé « T + très toxique », alors qu'il était jusque-là classé comme « nocif ». L'arrêté qui devait être pris pour en encadrer l'usage n'a pas été pris.
Le rapport d'expertise judiciaire de décembre 2003 confirme le lien entre la mortalité des abeilles et les risques qui restent à préciser pour l'homme. C'est à la suite de ces développements qu'intervient le 23 février 2004 la suspension de la vente des insecticides à base de fipronil, au premier rang desquels le Régent TS (de BASF) par le ministre de l'agriculture. Cette suspension est décidée jusqu'à l'achèvement de la procédure communautaire d'évaluation prévue au plus tard le 31 décembre 2005. Toutefois un délai a été accordé pour les stocks de semences enrobées pour les semis de printemps 2004 (environ 95 % des besoins).
Parallèlement, les directions générales des trois ministères de tutelle de l'AFSSA ; DGAL, DGCCRF et DGS ainsi que deux directions du ministère de l'écologie et du développement durable sont amenées à saisir l'AFSSA et l'AFSSE « des risques pour la santé humaine liés au fipronil ». L'explicitation de la saisine rappelle comment, après qu'il ait été autorisé, le fipronil est en quelque sorte « mis en examen » dans le cadre d'une évaluation pour l'AESA, pleinement compétente au niveau communautaire pour ce type de produits, et par les expertises rendues tout récemment dans le cadre de l'ordonnance judiciaire sur le problème des abeilles (Pr Arnold et Narbonne) :
« La Commission d'étude de la toxicité des produits antiparasitaires et assimilés (Comtox), instance compétente au niveau national en matière d'évaluation de la toxicité des pesticides, s'était prononcée sur les préparations à base de fipronil. Dans le cadre de la directive 91/414/CEE du 15 juillet 1991 concernant la mise sur le marché des produits phytopharmaceutiques et du règlement (CE) n° 451/2000 de la Commission du 28 février 2000 établissant des modalités de mise en oeuvre du programme de travail visé par la directive, elle a poursuivi ses investigations en vue de la transmission à l'Autorité européenne de sécurité des aliments (AESA) du projet de rapport d'évaluation de cette substance active pour laquelle la France est l'Etat membre rapporteur. Ce projet contient une recommandation visant à la non inscription du Fipronil à l'annexe I de la directive 91/414/CEE, pour des raisons liées à des risques pour l'environnement, et ce, en l'attente d'un débat contradictoire avec le pétitionnaire et les Etats membres.
Parallèlement, un rapport récent d'expert sollicité par ordonnance judiciaire pose de nouvelles interrogations sur les risques pour la santé. Il fait état dans sa conclusion « d'effets possibles sur l'homme à court terme par inhalation et à long terme par ingestion (dépassements possible de la dose journalière admissible) ». ces éléments, portés à la connaissance des pouvoirs publics, ne recouvrent pas les conclusions élaborées par la Comtox. Ces incertitudes scientifiques actuelles doivent être levées pour des impératifs de santé publique et il convient de s'intéresser à l'ensemble des usages possibles du fipronil ».
Concernant l'imidaclopride (substance de base du Gaucho), une « procédure contradictoire » auprès de Bayer Cropscience et des utilisateurs concernés a été engagée en même temps par le ministre de l'agriculture (l'arrêt du Conseil d'Etat sur le Gaucho interviendra cinq semaines plus tard).
Les freins, les dissimulations et les opérations de diversion n'auront donc pas réussi à empêcher que les problèmes graves que recelaient ces deux dossiers soient enfin traités plus normalement. Il a fallu, en ce qui concerne le Gaucho, deux annulations successives par le Conseil d'Etat, et de l'ouverture d'enquêtes judiciaires pour apporter une lumière crue sur des mécanismes administratifs qui ont d'évidence besoin d'être réformés. Cette réforme indispensable permettrait que la loi du 1 er juillet 1998 soit enfin appliquée dans le domaine des produits phytosanitaires, en liaison avec celle du 9 mai 2001 pour ce qui concerne l'AFSSE. La saisine commune du 27 février 2004 sur le fipronil cité ci-dessus indique en quelque sorte le chemin.
Au-delà de l'illustration factuelle que constituent les affaires du Gaucho et du Régent, il convient de signaler que les entités que sont l'AFSSA et la DGCCRF ne peuvent, dans la situation actuelle non réformée, jouer le rôle nécessaire qui leur revient. En outre, dans le secteur agricole, à l'extérieur de la DGAL, des remarques critiques se sont aussi fait entendre. Ainsi le service central des enquêtes du ministère de l'agriculture s'est étonné de la non publication d'une étude consacrée à l'utilisation des pesticides sur le blé et le maïs en 2001.
L'organisation actuelle est marquée par de graves faiblesses :
-- La compétence restreinte de la DGCCRF ne lui permet pas, par exemple, d'effectuer des contrôles sur les produits d'origine animale en ce qui concerne une éventuelle teneur en pesticide.
-- La compétence de l'AFSSA dans ce domaine des produits phytosanitaires est également très restreinte, résiduelle si l'on ose dire, car c'est précisément par la mise en cause de résidus de ces produits dans les aliments qu'elle peut être amené à en connaître. Elle participe également à l'instruction des projets de textes européens et a, de ce fait, à connaître des dispositions existantes et envisagées.
Sur le plan de l'expertise, les laboratoires ne peuvent pas coopérer comme le fonctionnement normal des différentes unités scientifiques et administratives l'exigerait. Ces difficultés ont trouvé une illustration exemplaire avec la situation dans laquelle s'est trouvé le laboratoire de l'AFSSA de Sophia Antipolis, spécialisé notamment sur les abeilles et qui est bien sûr sollicité dans les problèmes soulevés par le Gaucho et le Régent.
En effet, les scientifiques ne sont que très partiellement associés à l'élaboration des protocoles et en aucun cas à la synthèse des résultats obtenus auprès des différents intervenants ; ils n'ont pas, de ce fait, accès à l'ensemble des informations.
Lorsqu'une mesure de retrait de plusieurs lots de semences traitées avec du fipronil a été prise en septembre 2003 par le ministère de l'agriculture, probablement en s'appuyant sur des travaux auxquels l'Agence a contribué, les résultats de ces études ne lui ont pas été transmis, ni en sa qualité de laboratoire, ni d'évaluateur des risques.
Des améliorations en cours ?
En matière de sécurité sanitaire, la grave lacune observée dans le domaine des produits phytosanitaires a fait l'objet depuis plusieurs mois d'un constat de plus en plus partagé. Depuis les événements du printemps 2004, ce constat fait l'unanimité des acteurs administratifs.
Ainsi, après une série de rapports sur la présence de résidus de pesticides dans les milieux avec lesquels l'homme est en contact, il a été décidé le 27 novembre 2003, par les quatre ministres concernés (écologie, santé, agriculture, commerce et consommation) de mettre en place un « observatoire des résidus de pesticides », concrétisant ainsi un projet préparé depuis deux ans.
La mission première confiée à cet observatoire est :
-- de rassembler, en vue de leur valorisation, les informations et résultats des contrôles et mesures de résidus de pesticides dans les différents milieux et produits consommés par l'homme ;
-- d'estimer les niveaux d'exposition des populations ;
-- d'identifier les actions de progrès pouvant être mises en place sur les systèmes d'informations et notamment la nature et le format des données collectées.
L'observatoire des pesticides est une structure mise en place sous la forme d'un comité de pilotage constitué des administrations concernées (DGS, DGAL, DE, DGCCRF). Il associera l'AFSSE, l'AFSSA et l'IFEN de même que les autres instituts et agences publics concernés en tant que de besoin.
Dans ce cadre, en vue d'alimenter les travaux de l'observatoire, il a été demandé d'engager un travail préparatoire à l'évaluation de l'exposition des personnes. Dans un premier temps, les travaux permettront :
-- d'analyser les conditions nécessaires (cohérence des plans d'échantillonnage, types de mesures et leur validité, format des données, mode de stockage, possibilité de centralisation ...) pour que les différentes bases de données identifiées puissent être combinées pour produire des indicateurs d'exposition globale intégrant les différentes sources et voies d'exposition.
-- de proposer les indicateurs utiles à la caractérisation de l'exposition (milieux ambiants et milieu professionnel) de la population et à sa surveillance ainsi que les données utiles à leur construction.
-- de faire des propositions pour homogénéiser les données disponibles et le cas échéant les corriger.
-- d'identifier les pistes d'amélioration des systèmes d'information existants.
A partir de ces données de base, une première évaluation de l'exposition des populations devrait être réalisée pour la fin du premier semestre 2004.
Les pratiques administratives, le contrôle sur le terrain notamment, connaissent depuis 1999 et la loi d'orientation agricole, une évolution positive. Les découvertes récentes et nombreuses d'importations frauduleuses de produits interdits en attestent. Cela dit, ces découvertes ont quelquefois un caractère « providentiel » qui d'une certaine manière montre que l'administration avait connaissance de certaines pratiques. Or, les risques dans ce domaine augmentent extrêmement vite actuellement.
A cet égard, le recul opéré sur le contrôle des pulvérisateurs est parlant. Ce contrôle devait être mis en place en 2002 ; l'entrée en vigueur effective de ce dispositif comparable au contrôle technique des automobiles a été reportée et n'a toujours pas eu lieu. Cette mesure permettrait pourtant d'éliminer les matériels vétustes et inadaptés. Les premiers bénéficiaires en seraient évidemment les agriculteurs à un moment où les risques qui pèsent sur eux sont de plus en plus souvent soulignés.
Ce recul regrettable pourrait amener à conclure que si les risques pour les agriculteurs, l'environnement, les habitants et les consommateurs se révélaient être d'une gravité réelle, on serait amené à changer d'organisation.
L'examen du dispositif global actuel de traitement des récoltes et du contrôle des résidus montre des faiblesses à plusieurs niveaux (évaluation des LMR, bonnes pratiques agricoles, sécurité des applicateurs, volume des contrôles, risque environnementaux ...).
Pour combler les lacunes, des efforts considérables mettant en jeu des intérêts divergents vont être nécessaires.
La même remarque vaut pour l'ensemble de la question posée par les produits phytosanitaires. Les améliorations qui viennent d'être évoquées sont réelles, mais insuffisantes à elles seules pour redresser la situation. Et la création d'une nouvelle agence ne peut être une solution alors même qu'elle a été évoquée par le ministère de l'agriculture.
Une agence pour la santé des végétaux
Dans un développement chronologique qui ne semble rien devoir au hasard, M. Hervé Gaymard, ministre de l'Agriculture avait annoncé devant le congrès de la FNSEA le 1 er avril 2004 qu'il souhaitait « mettre en place rapidement une agence pour la santé des végétaux qui réunisse des scientifiques de divers horizons. Je pense en particulier à des spécialistes de la santé publique, à des agronomes, à des experts de la sélection végétale. Nous nous donnerons ainsi les moyens d'orienter de façon cohérente et pérenne notre agriculture vers des pratiques à la fois plus respectueuses de notre environnement et économiquement performantes ».
Intervenant le lendemain de l'arrêt du Conseil d'Etat annulant la décision du ministre autorisant le Gaucho, cinq semaines après l'inévitable décision de suspension du fipronil, cette proposition inattendue a-t-elle pour objet de répondre aux critiques ou de créer une instance qui permette d'éviter l'application de la loi de 1998 dans ce domaine en s'assurant ainsi que l'AFSSA, notamment, n'aura pas à connaître du fond des questions posées. Joker ou opération de diversion, l'Agence pour la santé des végétaux semblerait procéder du réflexe : un problème - une agence. Avec le même raisonnement, les produits de contact alimentaire auraient pu faire l'objet d'une agence à eux seuls, compte tenu des développements complexes que connaît ce secteur. Sauf à ignorer l'esprit et la lettre de la loi du 1 er juillet 1998, une telle création ne paraît ni cohérente, ni viable.
Cette idée n'ayant pas été développée depuis, et le poste ministériel ayant changé de titulaire, on peut estimer que cette proposition n'est sans doute plus d'actualité.
En revanche, une réelle coopération entre administrations concernées avec une mutualisation des expertises, des contrôles et des recherches constituerait certainement la base d'une réforme qui permettrait de conduire à son terme de la loi de 1998. Le fonctionnement d'une unité comme la SSM illustre la possibilité d'une expertise sans doute améliorable, mais tangible et digne de ce nom. D'ailleurs, le détail de la saisine de l'AFSSA et de l'AFSSE par les directions des quatre ministères (cf. supra) du 27 février 2004, illustre tout à fait la faisabilité d'une organisation et d'une démarche d'expertise dans le cadre d'une interministérialité inévitable ; la formule expérimentale ainsi choisie préfigure ce que pourrait être une architecture pérennisée :
« Aussi, nous vous demandons de bien vouloir constituer un panel d'experts commun, qui sera composé de membres émanant tant du comité d'experts spécialisés « Résidus et contaminants chimiques et physiques » de l'Agence française de sécurité sanitaire des aliments que du comité d'experts spécialisés « Evaluation des risques liés aux substances chimiques » de l'Agence française de sécurité sanitaire environnementale, ceci permettant d'associer, en toute impartialité, des personnalités scientifiques qualifiées particulièrement concernées par l'évaluation demandée.
Nous vous précisons, par ailleurs, que l'ensemble des pièces du dossier dont nous disposons, vous seront transmises notamment par le président de la Commission d'étude de la toxicité, M. Daniel Marzin, et par M. Thierry Mercier, Directeur de la SSM (INRA de Versailles), chargé du secrétariat scientifique de la Commission d'étude de la toxicité, afin que les personnalités qualifiées qui seront conduites à se prononcer, dans le cadre de cette expertise collective, puissent le faire en disposant de tous les éléments en notre possession ».
L'évaluation des produits phytosanitaires serait la tâche d'un comité d'experts commun à l'AFSSA et l'AFSSE, la structure scientifique mixte continuant à jouer dans ce cadre son rôle essentiel gardant son lien avec l'INRA, mais évidemment pas avec la DGAL ; cette dernière pourrait garder une simple représentation au sein du comité d'homologation pour l'évaluation de l'efficacité des produits. Ce dernier, s'il était maintenu, devrait avoir un positionnement très différent, satisfaisant quant à la transparence.
Après l'expertise, la décision doit intervenir dans les conditions claires et tenir compte du retour d'expérience qui doit lui-même être loyalement partagé entre les différentes entités concernées, contrairement à ce qui s'est passé encore récemment.
Le périmètre de compétences de l'AFSSA n'a donc pas à être modifié : en revanche les dispositions réglementaires devraient être prises afin que la lettre et l'esprit de la loi soient respectés pour l'ensemble des produits phytosanitaires ; il convient de préciser en outre que le respect de cette compétence de l'AFSSA sur la filière végétale implique le transfert de la Comtox à l'Agence.
2.5.2. Les OGM : un domaine trop partagé
L'évaluation des risques en ce qui concerne les O.G.M. est répartie, pour des raisons contingentes, entre différentes instances. Cette répartition n'exclut pas l'AFSSA, mais la met en concurrence, avec d'autres instances, dans des conditions qui ne sont pas satisfaisantes et donnent lieu à des appréciations contradictoires.
La répartition des expertises est ainsi définie :
-- la commission du génie génétique (CGG) crée en 1989, rattachée au ministère de la recherche se prononce sur l'utilisation d'OGM en milieu confiné ;
-- la commission du génie biomoléculaire (CGB), crée en 1993, placée auprès des ministres de l'agriculture et de l'environnement, chargée de l'évaluation des risques liés à la dissémination volontaire d'OGM à des fins de recherche ou de mise sur le marché (16 dossiers examinés en 2002, concernant uniquement des expérimentations, dont 10 concernant des plantes et 6 concernant des essais de thérapie génique).
-- et le comité d'experts spécialisés sur la biotechnologie de l'AFSSA qui se prononce sur les OGM destinés à l'alimentation (article l. 1323-2, 7° alinéa du code de la santé publique).
L'articulation entre les compétences de la CGB qui a un large domaine de compétences et celles de l'AFSSA à travers son comité d'experts spécialisés biotechnologies (appréciation des seuls risques liés à la consommation de produits alimentaires avant leur mise sur le marché) n'est pas assurée. Ces deux instances d'expertise peuvent donc être simultanément ou successivement être saisies de la même question ; les exemples ne sont pas rares et cette configuration explique que des avis contradictoires puissent être rendus. Cela a été le cas récemment avec le maïs MON 863.
La CGB donne le 28 octobre 2003 un avis au terme duquel, à la suite d'expérimentations d'alimentation menées sur le rat, elle estime qu'elle n'est pas en mesure de conclure à l'absence de risque pour la santé animale et humaine. L'AFSSA, également saisie, rend un avis opposé quelques jours plus tard le 6 novembre 2003 estimant que les différences observées entre les deux groupes sont « sans signification biologique » et que, le maïs MON 863 ne présente pas de risque nutritionnel ». L'Autorité européenne conclut à son tour le 19 avril 2004 en donnant un avis favorable considérant que les différences enregistrées rentrent dans les variations normales des populations de contrôle. Outre le chevauchement des zones de compétence entre la CGB et l'AFSSA, apparaît une nouvelle interférence entre cette dernière et l'Autorité européenne.
Un autre cas s'est également posé récemment avec le maïs Bt 11. En avril 2002, le comité scientifique sur l'alimentation au niveau européen a donné un avis favorable estimant « qu'il n'y a pas d'indication que le maïs doux Bt 11 est moins sûr pour la consommation humaine qu'un maïs non transgénique ». L'AFSSA, rendant son propre avis le 3 décembre 2003, indique qu'afin d'éliminer des risques métabolique éventuellement liés à ce maïs spécifique, « il conviendrait d'évaluer l'impact d'une consommation régulière de celui-ci « considérant que les conclusions acquises sur le maïs réservé à l'alimentation d'élevage ne sont pas suffisantes ».
Deux dispositions européennes apportent encore un élément de complication supplémentaire. D'une part, la directive 2001/18 du 12 mars 2001 relative à la dissémination volontaire d'OGM dans l'environnement dont le délai de transposition était fixé à octobre 2002 et pour laquelle un avant projet de loi est en cours de préparation. D'autre part, le règlement européen du 22 septembre 2003 sur les denrées alimentaires et les aliments pour animaux génétiquement modifiés au terme duquel l'évaluation du risque est faite par l'Autorité européenne (AESA) qui peut demander aux agences nationales de s'assurer de l'innocuité alimentaire, mais aussi à la CGB (autorité compétente selon la directive 2001/18) pour évaluer le risque environnemental.
Le fonctionnement et la composition mêmes de la CGB sont problématiques au regard des principes posés par la loi de 1998 quant à la séparation entre l'instance d'évaluation et celle de gestion du risque. En effet, son secrétariat est assuré par la DGAL du ministère de l'agriculture avec la contribution du ministère de l'écologie. Par ailleurs, des représentants de la « société civile » sont prévus dans une commission qui est une instance d'expertise. L'atmosphère qui caractérise les débats sur les OGM n'y est peut-être pas étrangère ; il n'en reste pas moins que cette composition ne correspond pas aux principes d'organisation retenus par la loi.
Une clarification débouchant sur une restructuration s'impose donc. Comme on l'a envisagé pour les produits phytosanitaires, il convient d'une part de tirer les conclusions logiques de la séparation voulue entre l'évaluation et la gestion du risque, la DGAL n'intervenant plus dans le fonctionnement du dispositif, et d'autre part d'unifier en une seule commission ou comité d'experts, les deux instances actuellement existantes, en la plaçant conjointement auprès de l'AFSSA et de l'AFSSE.
Enfin, le lien prévu pour l'avant-projet de loi avec la surveillance des produits phytosanitaires à travers le comité de vigilance (art. L 125-1 du code rural) paraît en toute hypothèse à exclure.
2.5.3. Les allégations santé et les compléments à base de plantes : des compétences frontalières difficiles
Il s'agit ici de produits spécifiques dont l'importance peut paraître marginale si l'on s'en tient aux quantités en cause. Les questions qu'ils soulèvent sont d'une moindre ampleur que celles portant sur les produits phytosanitaires ou les OGM. Mais elles posent des questions totalement nouvelles qui justifient pleinement un examen particulier. Retenons un point de départ simple : pour l'essentiel, les conditions dans lesquelles elles sont traitées sont satisfaisantes, alors que la configuration des compétences et des circuits administratifs est complexe.
2.5.3.1. Les allégations santé des produits alimentaires
L'AFSSA et l'AFSSAPS ont ici des compétences partagées alors que la DGCCRF est en première ligne. C'est en effet au titre du code de la consommation (art. L 121-2) que les mentions alléguant d'un bénéfice pour la santé doivent pouvoir être prouvées scientifiquement. La DGCCRF, en charge de l'application et du contrôle, saisit donc l'AFSSA en vue d'expertise, laquelle est confiée au comité d'experts en nutrition humaine.
Parallèlement, les dispositions du code de la santé publique (art. L 5122-14) s'imposent : lorsque la publicité affirme, suggère ou suppose qu'un produit possède des propriétés curatives ou préventives à l'égard des maladies humaines ou des effets de restauration, correction ou modification de fonctions organiques, les producteurs ou distributeurs doivent soumettre une demande de vise à la commission administrative dite « du visa PP » (publicité produit) dont le secrétariat est assuré par l'AFSSAPS.
L'AFSSA et l'AFSSAPS ont établi une procédure conjointe de traitement pour garantir la cohérence du traitement de ces dossiers : l'AFSSA mène l'évaluation scientifique des dossiers déposés au titre du visa PP ; un représentant del'AFSSAPS se rend aux réunions du CES nutrition lorsque les dossiers d'allégations sont examinés ; un représentant de l'AFSSA se rend aux réunions préparatoires de la commission du visa PP.
Cette procédure, qui est bienvenue, ne permet pas néanmoins de résoudre les problèmes de fond qui se posent ici. Les entreprises peuvent engager le processus à différents niveaux, le cadre administratif de ces produits est éclaté. Il n'y a pas de référentiels scientifiques généraux d'évaluation sur les allégations santé même si l'AFSSA a publié des lignes directrices générales sur les dossiers industriels soumis au comité d'expert spécialisé « nutrition », et si un projet de règlement est à un stade d'élaboration avancé.
Par ailleurs, l'impossibilité juridique et matérielle de contrôler l'absence de demandes de visa affaiblit la portée du dispositif « publicité produit ». Quels que soient les efforts de la DGCCRF pour les contrôles qu'elle exerce effectivement, l'absence de tout mécanisme d'autorisation ou de déclaration ne permet pas un recensement, ni même une identification tangible du recours aux allégations. Les différents interlocuteurs interrogés à ce sujet ont tous fait part de l'insuffisance des réglementations, et notamment le Conseil national de l'alimentation. Il apparaît que le nombre de dossiers soumis à l'AFSSA est beaucoup trop faible et qu'une modification de la réglementation devrait s'attacher à rendre systématique la vérification des allégations.
En ce qui concerne la publicité, l'effectivité du rôle du BVP (Bureau de vérification de la publicité) a directement été mise en cause par tous les interlocuteurs interrogés.
Une amélioration de la situation est envisageable à partir d'une proposition de règlement européen concernant les allégations nutritionnelles et de santé. Ce texte, qui devrait entrer en vigueur en 2005 prévoit que l'usage d'allégations de santé dans l'étiquetage, la présentation et la publicité de denrées alimentaires sera soumis à une procédure d'autorisation, après évaluation scientifique par l'Autorité européenne de sécurité alimentaire (AESA). Si l'AESA saisit les agences nationales sur ces dossiers, l'AFSSA pourra procéder à une évaluation des allégations sur leur validité scientifique et leur formulation (sur le produit ou par la publicité).
Il serait souhaitable d'unifier la procédure en la confiant à l'AFSSA. A défaut, il est urgent et nécessaire de donner aux services de contrôle les moyens juridiques et administratifs d'intervenir efficacement dans ce domaine des « allégations santé ». En quelques années, ce qui n'était qu'une question marginale est en passe de devenir un réel problème car il faut bien admettre que des producteurs ou distributeurs très imaginatifs n'ont guère de difficulté à aller à la rencontre de publics avides de produits « miraculeux » en croyant être averti des réalités scientifiques.
C'est d'ailleurs la même tendance qui se retrouve pour les compléments alimentaires à base de plantes.
2.5.3.2. Les compléments alimentaires à base de plantes
L'AFSSA, mais aussi l'AFSSAPS intervenant dans ces domaines, les deux agences ont associé leurs personnels scientifiques et les experts des comités pour assurer la cohérence de l'évaluation de ces produits. Le rapport « Démarche pour l'évaluation de la sécurité, de l'intérêt et de l'allégation des denrées alimentaires à base de plantes pour l'alimentation humaine » a aussi été produit en collaboration. Ici aussi la vigilance face à certaines évolutions de consommation s'impose. On rejoint dans ce cas des problèmes qui relèvent de la pharmacie et plus exactement de l'herboristerie; certains témoignages d'interlocuteurs britanniques illustrent les risques que pour des plantes inconnues ou mal connues, la multiplication des points d'entrée y compris par internet, fait courir.
La collaboration satisfaisante entre l'AFSSA et l'AFFSSAPS s'est également illustrée avec le cas des aliments diététiques destinés à des fins médicales spéciales. Sans entrer dans le détail de la réglementation, les textes répartissent les compétences entre elles. Des améliorations restent à apporter, notamment quant à l'échange d'informations sur les dossiers évalués (composition, étiquetage).
2.6. Les laboratoires : une nécessité et des clarifications
2.6.1. Une configuration décidée par le législateur
Lors de l'examen de la loi du 1 er juillet 1998, le législateur s'est décidé en faveur de l'intégration de l'ensemble du CNEVA (Centre national d'études vétérinaires et alimentaires), au sein de la nouvelle agence, l'AFSSA. Cette décision a été précisée dans les termes suivants (art. L 1323-1 du code de la santé publique : « Dans le cadre du Centre national d'études vétérinaires et alimentaires, placé en son sein et géré par elle, l'agence fournit l'appui technique et scientifique nécessaire à la mise en oeuvre des mesures prévues par le code rural.
Pour l'accomplissement de ses missions, les laboratoires des services de l'Etat chargés du contrôle de la sécurité sanitaire des aliments et ceux qui leur sont rattachés sont mis à disposition de l'agence en tant que de besoin. (anc. Art. L. 794-1, I) ».
Cette intégration, dont le caractère « contingent » a été souvent souligné (cf. infra) a permis de constituer au sein de l'AFSSA un ensemble de laboratoires qui, dès le début du fonctionnement de la nouvelle agence, lui a donné les moyens nécessaires aux missions d'appui scientifique et technique et aux tâches de recherche qui lui ont été fixées, au-delà de l'évaluation des risques sanitaires des aliments.
En effet, la loi a explicitement énoncé que l'agence devait exercer des missions de recherche puisque l'article L. 1323-2, 5° du code de la santé publique dispose que l'Agence française de sécurité sanitaire des aliments : « mène, dans le respect du secret industriel, des programmes de recherche scientifique et technique dans les domaines du génie vétérinaire, de la santé animale, du bien-être des animaux et de leurs conséquences sur l'hygiène publique, ainsi que de la sécurité sanitaire des aliments. A cette fin, elle mobilise ses propres moyens ou s'assure le concours d'organismes publics ou privés de recherche ou de développement, d'universités ou d'autres établissements d'enseignement supérieur, de collectivités territoriales ou de personnes physiques ».
De plus, le décret du 26 mars 1999 prévoit que le conseil scientifique de l'agence « concourt à la définition de la politique nationale de recherche en matière de sécurité sanitaire des aliments. A cet effet, il peut formuler des recommandations sur toute question scientifique et technique entrant dans le champ de compétence de l'établissement. Celles-ci sont transmises au directeur général et au Président du conseil d'administration ».
L'AFSSA est donc en charge de fonctions d'appui scientifique et technique et de missions de recherche, en particulier à travers les laboratoires dont elle a été dotée, sans qu'il y ait une coïncidence exacte entre le champ de compétence de l'Agence dans ces domaines et celui qui est le sien en matière d'évaluation des risques.
En outre, les activités relatives à la santé animale ont une place particulièrement développée, compte tenu notamment du fait que le CNEVA, a été intégré à l'Agence dans sa totalité. L'existence au sein de l'AFSSA d'un directeur chargé de la santé et du bien-être des animaux, nommé par arrêté du ministre de l'agriculture, sur proposition du directeur général, illustre cette particularité.
Aux laboratoires du CNEVA s'est ajouté le laboratoire d'hydrologie situé à Nancy qui lui relevait jusque-là de la DGS (Direction générale de la santé) et qui a été intégré directement à la structure de l'AFSSA. Les treize laboratoires de l'AFSSA sont répartis sur dix sites dont celui de Maisons-Alfort qui en compte trois. Sur les 900 agents de l'AFSSA, 684 soit 76 %, sont affectés aux laboratoires.
Cette situation particulière comporte des atouts pour l'Agence, et c'était le but recherché, même si la décision a été prise sans préparation à une très faible majorité (une voix au Sénat), et fortement discutée avant l'adoption définitive. Elle comporte aussi des contraintes qui, l'expérience le montre, sont gérables. L'AFSSA caractérise d'ailleurs elle-même cette situation dans son document « La recherche, missions et politique de l'AFSSA ; orientations 2002/2005 » :
« La coexistence de missions générales d'évaluation des risques, de missions de recherche et des missions d'appui scientifique est une originalité dans le dispositif sanitaire français : l'Agence française de sécurité sanitaire des aliments est la seule agence issue des modifications législatives des dix dernières années à avoir la majorité de ses moyens et de ses effectifs situés dans des laboratoires qui accomplissent des activités de recherche et d'appui scientifique et technique.
Si cette originalité peut être source de difficultés, liées à la conciliation de fonctions différentes au sein d'un même établissement, elle représente également un atout essentiel qu'il convient de valoriser au mieux et de conforter. Elle permet en effet d'instaurer des interactions réciproques entre ces différents métiers de manière sans doute plus rapide, plus souple et plus fructueuse que s'ils étaient exercés dans des institutions différentes.
A l'appui de cette vision, il faut souligner qu'à l'étranger, d'autres organismes ayant des responsabilités dansle champ de la sécurité sanitaire ont développé des activités de rechercher dans leurs laboratoires (c'est le cas notamment de la FDA aux Etats-Unis) ou disposent de moyens spécifiques pour développer la recherche dans d'autres organismes (c'est le cas par exemple de la Food Standards Agency en Grande-Bretagne). (...)
Il convient enfin de souligner que les activités de recherche peuvent être un moyen privilégié de maintien d'un bon niveau scientifique pour d'autres activités. C'est ainsi que dans son analyse du fonctionnement du laboratoire d'hydrologie avant son rattachement à l'AFSSA par le décret du 26 mars 1999, l'Inspection générale des affaires sociales avait déploré que ce laboratoire se soit progressivement coupé des problématiques de recherche dans son domaine de compétence et avait évoqué le retentissement de cette situation sur la qualité des autres activités du laboratoire ».
Ces observations sont d'autant plus intéressantes que dans la perspective de clarifications inévitables, les configurations les plus diverses, quelque peu éloignées de ce tableau, sont avancées.
2.6.2. Un appareil tangible mais complexe
L'intégration du CNEVA, donc entre autres de la totalité des structures propres à la santé animale, a été plutôt bénéfique, voire très bénéfique. Les avis sont concordants. Contrairement aux craintes exprimées lors des débats de 1997-98 et pendant la période de mise en place, on s'accorde à reconnaître que la santé animale n'a pas du tout été négligée. La nouvelle agence a maintenu les acquis hérités au CNEVA et a même procédé à quelques développements. Les ressources n'ont pas pâti de cette intégration, au contraire ; les relations avec l'industrie ont été clarifiées, même si certains inconvénients sont ressentis par rapport à des coopérations qui ne peuvent plus s'établir autant que certains le souhaiteraient.
L'intégration et l'encadrement des laboratoires ont été illustrés par des progrès significatifs et des initiatives innovantes :
-- Développement des réseaux de surveillance épidémiologique ; ces réseaux - il convient de le souligner - étant gérés conjointement par l'AFSSA et la DGAL.
-- Propositions d'orientations des activités d'appui scientifique et technique, issues d'une analyse menée pendant deux ans par l'ensemble des scientifiques de l'Agence, le conseil scientifique, la DGAL ayant été associée.
-- Définition des orientations de recherches prioritaires 2002-2005, après inventaire dans les différents laboratoires. Cet inventaire réalisé pour la première fois pour l'ensemble des laboratoires de l'Agence a été utile à différents niveaux. Il a permis :
-- L'identification des domaines couverts et non couverts
-- La répartition des personnels scientifiques par thème et par axe stratégique
-- La définition de 13 axes de recherche
-- L'identification de thèmes et d'axes stratégiques prioritaires à renforcer
-- Les 3 axes stratégiques prioritaires retenus :
-- Les problèmes des filières bovines
-- La recherche dans le domaine de la pêche
-- La problématique liée à l'eau et aux végétaux
Les 5 thématiques prioritaires :
-- Les maladies émergentes et leur potentiel de transmission à l'homme
-- Les méthodes d'estimation qualitative des risques chez les consommateurs
-- L'alimentation animale et le risque pour l'homme
-- Les ESST
-- La composition des aliments et les risques nutritionnels
L'initiative importante concrétisée au niveau européen dans le cadre du 6è PCRD avec Med.Vet.Net. (prévention et contrôle des zoonoses transmissibles à l'homme) mérite d'être particulièrement remarquée. L'AFSSA est à l'origine du projet et en assure la coordination scientifique et administrative ; cette réalisation commune a été engagée avec quatre autres institutions intéressées (en Grande-Bretagne, Suède, Danemark, Pays-Bas) et s'étend maintenant à 16 institutions réparties dans 10 pays.
Des insuffisances ou des lenteurs dans la direction des laboratoires sont toutefois perceptibles en ce qui concerne la coordination des travaux et les accréditations. En outre, des choix ponctuels face à des problèmes que rencontrent deux unités sont contestables, le cas de l'abandon de la recherche en virologie bovine à Lyon étant le plus marquant.
Il faut souligner que les centres de recherche de l'AFSSA prennent place dans un dispositif plus large.
A côté des laboratoires de l'AFSSA, la santé animale est évidemment l'objet de travaux dans d'autres structures dont la finalité est précisément exclusivement la recherche. Ainsi en est-il avec l'INRA dont le département « santé animale » compte un effectif de 700 personnes, dont la moitié de scientifiques, répartis dans 23 unités sur neuf sites ; d'autres éléments de l'INRA (microbiologie), intervenant également dans le domaine animal.
Par ailleurs, le CIRAD (centre international de coopération internationale en recherche agronomique pour le développement) intervient aussi dans ce domaine par son département élevage et médecine vétérinaire tropicale (180 personnes).
Les laboratoires vétérinaires départementaux
Dans le domaine du contrôle et de la surveillance, les 78 laboratoires de contrôle (2500 personnes environ) réalisent la majorité des analyses rentrant dans ce cadre pour le compte de la DGAL. L'AFSSA rencontre ici de sérieuses difficultés pour obtenir la transmission des analyses réalisées par ces laboratoires, malgré des relations correctes avec les laboratoires de l'AFSSA. C'est là un problème évoqué par ailleurs et qui illustre la nécessité d'une réelle harmonisation des relations entre l'AFSSA et la DGAL dans l'usage commun des plans de contrôle et de surveillance.
Les laboratoires des ministères de tutelle : DGAL et DGCCRF
-- On rappellera au préalable que le laboratoire dépendant de la troisième tutelle qui est le ministère de la santé (à travers la DGS), à savoir le laboratoire d'hydrologie de Nancy, essentiellement centré sur le contrôle des eaux minérales, a été intégré à l'AFSSA en 2001. Un élargissement de ses compétences à toute la filière eau est actuellement souhaité par l'Agence et la DGS.
-- Le laboratoire national de la protection des végétaux compte un effectif de 450 personnes (dont un peu plus d'une centaine dans les laboratoires). Intégré dans la structure de la DGAL, il relève directement de sa sous-direction protection des végétaux. Il n'a pas été établi de lien entre lui et l'AFSSA.
-- La direction des laboratoires de la DGCCRF coiffe huit laboratoires sur l'ensemble de la France comptant au total 363 agents. La collaboration avec l'AFSSA est à développer même si les difficultés rencontrées dans la transmission des plans de contrôle et de surveillance n'est pas du même ordre qu'avec la DGAL.
2.6.3. La répartition des laboratoires : une nécessité confirmée
2.6.3.1. De multiples hypothèses
Compte tenu des difficultés que l'éparpillement des laboratoires fait naître, notamment pour l'Agence elle-même et sachant que tout schéma complexe suscite nécessairement des appétits de simplification, les propositions ne manquent pas. Ont ainsi été envisagées :
-- Concernant directement les laboratoires de l'AFSSA, le transfert pur et simple à d'autres organismes dont ceux centrés sur la recherche, à commencer par l'INRA.
-- Une redéfinition de la répartition thématique avec des établissements à caractère scientifique et technique : transferts des laboratoires n'ayant pas de compétences en matière de sécurité des aliments vers d'autres organismes ; transfert à l'AFSSA de laboratoires de recherche en sécurité des aliments venant de l'extérieur ;
-- Un regroupement des laboratoires ayant des compétences d'appui scientifique et technique (AFSSA, DGCCRF, protection des végétaux de la DGAL) au sein de l'Agence ou dans un autre organisme avec une compétence couvrant l'ensemble des champs de l'appui scientifique et technique : produits animaux, santé animale, médicament vétérinaire, produits végétaux, produits phytosanitaires, eau.
-- Une réorganisation des laboratoires en charge des missions de référence dans le secteur de la sécurité alimentaire avec la mise en place d'un double réseau : un réseau de centres de référence pour les maladies humaines sous la responsabilité de l'Institut de Veille Sanitaire et un réseau de référence pour la surveillance des aliments, de l'eau, des maladies animales sous la responsabilité de l'AFSSA.
La suppression de la distinction entre l'évaluation et la gestion du risque et en confiant cette dernière à l'AFSSA, amènerait à intégrer l'ensemble des laboratoires de la DGAL et de la DGCCRF à l'AFSSA. Dès lors que l'on décline cette orientation, une telle configuration ne peut être envisagée.
2.6.3.2. L'inévitable répartition
Lors de la journée d'auditions organisée en mai 2000 par la commission des affaires sociales du Sénat, un an après la mise en place du dispositif de la loi du 1 er juillet 1998, plusieurs échanges avaient permis de rappeler les nécessités d'organisation et d'articulation dans ce domaine de la sécurité sanitaire des aliments entre l'évaluation et la gestion du risque en soulignant l'élément essentiel qui est le positionnement des laboratoires. L'analyse faite alors par Mme Marylise Le Branchu, ministre en charge de la DGCCRF garde toute sa pertinence même si les « mise à disposition » de l'AFSSA doivent faire l'objet d'une sérieuse mise à niveau pour devenir effective en toute circonstance :
« Mme Marylise Le Branchu : Par rapport à ce que vous avez dit sur les deux agences, qui pourraient d'ailleurs être trois agences, puisqu'il s'agit d'environnement également maintenant, je vais essayer de regrouper les questions pour être plus brève.
J'ai l'intime conviction que l'on travaille dans un pays par étape. On ne coule pas dans le marbre une fois pour toutes des outils. Compte tenu du caractère récent de cette volonté d'avoir une évaluation transparente du risque, il n'est pas mauvais de séparer les fonctions. Il est probable, possible, que, dans quelques années, nous soyons amenés à revoir la structure des agences. Je pense qu'il faut d'abord faire fonctionner ce que nous sommes capables de faire fonctionner.
La co-tutelle nous a semblé être une nécessité au départ dans la mesure où nous voulions continuer à affirmer un principe fort : celui de la séparation absolue de l'évaluation et de la gestion du risque. A-t-on le même outil pour évaluer et gérer le risque ? A mon avis, non. Les organismes de contrôle doivent applique le résultat de l'évaluation du risque, prendre les processus d'évaluation qui leur ont été donnés et, pendant qu'ils contrôlent, l'Agence évalue d'autres risques potentiels, etc ... C'est un mouvement continu.
Les comités d'experts vous préoccupent. La loi sur l'Agence dit : « Pour l'accomplissement de ses missions, les laboratoires des services de l'Etat chargés du contrôle de la sécurité sanitaire des aliments et ceux qui leur sont rattachés sont mis à disposition de l'Agence en tant que de besoin ». Un premier mouvement a été décidé : le CNEVA n'est plus un laboratoire d'un service, mais est devenu un service de l'Agence, donc indépendant et agissant en transparence. Sa relation n'est que budgétaire. Pour les laboratoires, les résultats sont donnés à l'Agence pour ses évaluations constitutives. Elle a à sa disposition, quand elle veut formuler une question, tous les laboratoires de l'Etat.
Avoir une seule entité impliquerait une supra structure avec à l'intérieur tous les laboratoires, le tout en étant au service de l'évaluation du risque. Je reste persuadée qu'il faut garder des outils au service de la gestion du risque. L'Agence n'a pas comme vocation de contrôler. Elle perdrait son temps et son efficacité. En effet, le souci, que vous avez bien développé, était d'avoir un outil structurel institutionnel garanti transparent travaillant en permanence sur l'évaluation des risques potentiels. Laissons la gestion aux administrations de telle manière que l'Agence ne soit pas encombrée de missions de contrôle et ne fasse plus de travail d'évaluation aussi souvent qu'elle le pourrait.
Par ailleurs, il me semble que l'Agence n'aura jamais tous les laboratoires sur toutes les compétences dans l'avenir. Qu'elle ait le droit, en tant que de besoin, d'avoir à sa disposition ces laboratoires et que ceux-ci soient obligés de lui répondre sur toutes les évaluations qu'elle demande, y compris les laboratoires universitaires - des universités travaillent sur de la recherche fondamentale qui, à un moment donné, peuvent être utiles à l'Agence - est un fonctionnement qui me paraît devoir être poursuivi ; sinon, l'Agence manquera de moyens ».
2.6.3.3. Les besoins des services de contrôle
La nécessité pour les services en charge des contrôles de disposer de laboratoires s'impose. Cette question préalable ne constitue pas le seul argument qui justifie la répartition des laboratoires entre différentes entités administratives. Dès lors, c'est cette répartition qui doit être organisée, mieux organisée. Les doublons (dans le domaine de la santé animale par exemple) ne devraient pas perdurer et surtout les mécanismes qui imposent la collaboration entre administrations sont réalisables.
-- Par ailleurs, le transfert des laboratoires de l'AFSSA, soit à d'autres structures administratives existantes, soit à une structure nouvelle qui serait à créer, étant exclu, deux orientations complémentaires peuvent être envisagées ; elles constituent la voie d'une amélioration de l'outil créé par la loi de 1998 aux objectifs fixés. L'une consiste à procéder à des retouches, à des changements de rattachement, l'expérience enregistrée étant souvent le meilleur critère, l'autre consiste à instaurer des mécanismes de coopération entre administrations ou à les étendre lorsqu'ils existent déjà.
? Le transfert du Laboratoire national de protection des végétaux du ministère de l'agriculture à l'AFSSA apparaît comme une réponse envisageable et donc souhaitable face aux difficultés notées précédemment, particulièrement dans le domaine des produits phytosanitaires. L'appui scientifique et technique pour les fonctions de surveillance et de contrôle devrait naturellement continuer à être assuré au ministère de l'agriculture.
? L'absence de contrat d'objectifs et de moyens entre l'AFSSA et les administrations de tutelle a été pointée comme une gêne, sinon un obstacle, à une harmonisation des relations entre ces entités. Il est maintenant devenu une condition préalable à toute clarification, y compris pour l'Agence elle-même ; cela est d'autant plus urgent que la perspective de concrétisation de la LOLF s'approche. Mais pour le ministère de l'agriculture, ce préalable doit être complété par l'établissement de rapports d'ensemble dans le cadre d'une convention générale ; les réformes à opérer ne retireront pas au ministère de l'agriculture sa fonction de tutelle et celle de partenaire de l'Agence qui doivent être clairement distinguées.
Les laboratoires de la DGCCRF, de par leur positionnement et leur mission de contrôle ne soulèvent pas à notre sens de questionnement quant à leur rattachement ou à leur structuration. En revanche, des améliorations notables devraient être apportées dans le cadre d'une convention générale dont on peut s'étonner qu'elle n'ait pas encore été rédigée alors que les domaines d'action sont pour le moins connexes.
Les sujets concernés pourraient être, d'après les propositions de l'AFSSA : les programmes de surveillance des résidus de pesticides, les programmes de surveillance des résidus d'antibiotiques, les programmes de surveillance phytotoxines/mycotoxines, la recherche de la présence d'OGM et les résultats des contrôles de l'eau.
2.6.3.4. Les besoins de l'AFSSA pour la recherche
Une clarification dans les rapports avec le ministère de l'agriculture s'impose également en ce qui concerne la recherche. En effet, les tutelles ne jouent pas actuellement un rôle consistant dans l'orientation de la recherche au sein de l'AFSSA, ce qui laisse à celle-ci une marge d'appréciation, donc de choix, plutôt large. Or l'Agence ne peut revendiquer, tout au moins à elle seule, l'orientation de la recherche car elle est à la fois juge et partie. Les priorités liées à son « coeur de métier », l'évaluation du risque, peuvent au mieux être appréciées par elle, mais pour tout le reste, c'est différent. L'analyse faite par l'Agence elle-même des choix de la recherche dans le document précité 28 ( * ) ne manque pas de pertinence et mérite d'être citée, mais elle montre aussi la nécessité d'une implication plus grande du ministère dans l'orientation de ces choix :
« Les champs de la recherche
Pour aborder la question des champs de la recherche, deux approches sont possibles : l'une qui partirait des compétences actuelles des laboratoires ; l'autre qui analyserait les besoins généraux dans l'ensemble du champ de compétence de l'agence. Réflechir à une stratégie, c'est croiser ces deux approches, en déterminant comment les compétences actuelles de l'agence peuvent contribuer à répondre à des besoins de recherche.
C'est en partant des activités actuellement réalisées au sein des laboratoires, en renforçant les logiques transversales, qu'il convient d'examiner dans quels secteurs il serait nécessaire d'en faire plus ou moins, en se fondant sur les principes suivants :
-- une relation étroite entre secteurs de recherche et d'appui scientifique et technique et secteurs d'évaluation est souhaitable ;
-- compte tenu de son rôle et de sa mission de service public, il n'est pas envisageable que l'agence interrompe des activités considérées comme répondant à un besoin qu'elle est la seule ou la mieux à même de satisfaire ;
-- par symétrie, il est important que l'agence conserve une capacité de développer des activités de recherche répondant à un besoin non satisfait et renforcer une capacité d'adaptation à des thématiques émergentes ;
-- compte tenu de sa taille, le développement des activités de recherche de l'agence ne peut se faire que sur des objectifs précis.
L'autre approche consiste à partir de l'examen des missions de l'Agence pour identifier les quatre champs majeurs dans lesquels on pourrait considérer que l'AFSSA se doit de réaliser des recherches. Il s'agit :
-- des risques sanitaires des aliments destinés à l'homme et aux animaux d'élevage (y compris les eaux de boissons), ces risques pouvant résulter de contaminations microbiennes ou chimiques, délibérées (produits de traitements) ou fortuites (polluants) ;
-- de la valeur nutritionnelle et organoleptique des aliments destinés à l'homme ;
-- de la santé des populations animales élevées ou exploitées par l'homme, la notion de santé intégrant non seulement les pathologies, mais aussi les aspects liés aux conditions d'élevage et au bien-être animal ;
-- des risques sanitaires pour l'homme liés à la faune sauvage ou domestique, même lorsqu'ils ne constituent pas un problème direct de santé animale (phénomènes de portage) ou lorsque la transmission n'implique pas forcément un vecteur alimentaire (cas de la rage par exemple).
Dans la mesure où l'agence n'a pas vocation à couvrir l'ensemble des sujets de recherche dans ces différents domaines, elle se doit de stimuler des recherches dans des thèmes jugés prioritaires et pour lesquels il apparaît nécessaire de favoriser la complémentarité entre organismes.
C'est donc cette double démarche de réflexion que l'agence s'est proposée de mettre en oeuvre ».
2.7. Une agence dans l'Agence : l'Agence nationale du médicament vétérinaire
2.7.1. Histoire de l'ANMV
Issue d'un long processus qui a commencé avec la création à Fougères du laboratoire des médicaments vétérinaires en 1975, l'Agence nationale du médicament vétérinaire a été créée en 1994, puis intégrée à l'AFSSA en 1999 lors de la mise en place de celle-ci tout en gardant ses structures, son organisation et ses compétences. Son directeur est l'un des directeurs placé auprès du directeur général de l'AFSSA, mais il est nommé, sur proposition de ce dernier, par arrêté des ministres chargés de l'agriculture et de la santé ; l'Agence nationale du médicament vétérinaire est placée sous la tutelle conjointe de ces deux ministres alors que l'AFSSA elle-même, dont elle constitue un élément, est sous la triple tutelle déjà décrite (la troisième étant celle du ministre chargé de la consommation à travers la DGCCRF).
Compte tenu du caractère particulier de sa mission spécifique sur le médicament vétérinaire, l'ANMV a des compétences étendues et précisées dans le code de la santé publique à travers le directeur général de l'AFSSA ; l'Agence est chargée : « d'assurer auprès du ministère de l'agriculture et des autres ministres intéressés l'appui scientifique et technique nécessaire à l'élaboration, à l'application et à l'évaluation des mesures prises dans les domaines de la santé animale, du médicament vétérinaire, du bien-être des animaux et de leurs conséquences sur l'hygiène publique, ainsi que la sécurité sanitaire des aliments destinés à l'homme ou à l'animal. Elle participe au contrôle de l'utilisation et de la dissémination des organismes génétiquement modifiés, en ce qui concerne les médicaments vétérinaires dans les conditions prévues parle décret n° 95-1173 du 6 novembre 1995 pris pour l'application du titre III de la loi n° 92-654 du 13 juillet 1992 relative au contrôle de l'utilisation et de la dissémination des organismes génétiquement modifiés, en ce qui concerne les médicaments vétérinaires » (art. R. 794-2 du code de la santé publique).
A ce titre, le directeur de l'AFSSA, est chargé non seulement de l'évaluation du médicament vétérinaire, mais également de mesures de gestion au nom de l'Etat par les articles L 1323-5 et R 794-16 (autorisation des médicaments vétérinaires et des établissements pharmaceutiques vétérinaires, activités d'inspection et de contrôle par exemple). Par contre, les mesures de police sanitaire à l'égard des médicaments non soumis à autorisation préalable relèvent toujours des ministres chargés de l'agriculture et de la santé, à la différence de l'AFSSAPS pour le médicament à usage humain.
Les missions confiées à l'AFSSA relevant de l'ANMV visent :
-- les autorisations de mise sur le marché de ces médicaments, ainsi que d'autres décisions qui y sont plus ou moins liées : autorisations des essais cliniques, autorisations temporaires d'utilisation, autorisations des autovaccins, autorisations d'importation, certifications à l'exportation ;
-- la pharmacovigilance, le contrôle de la publicité et de la qualité des médicaments sur le marché ;
-- les autorisations relatives à différentes catégories d'établissements : fabrication, importation, exportation, distribution en gros de médicaments vétérinaires, fabrication d'aliments médicamenteux ;
-- l'inspection de ces établissements ;
-- l'appui à la conception de textes législatifs et réglementaires.
L'organisation des travaux de l'ANMV s'est donc structurée depuis longtemps et ce, autour de ces compétences bien précises. Les différents services ou comités techniques fonctionnent et selon des schémas adaptés et clairs. Il y a lieu de noter que l'ANMV est, après ses homologues britannique et allemand, la troisième entité en Europe en terme d'effectifs.
2.7.2. Les effets de l'intégration
Les effets de l'intégration de l'ANMV dans l'AFSSA ne paraissent pas avoir été très tangibles ; les avantages qu'elle aurait pu y trouver restent encore à prouver.
On peut certes considérer qu'on n'a pas en sens inverse enregistré d'inconvénients notoires, mais cela reste peu satisfaisant même si l'ANMV s'acquitte d'une mission essentielle dans le cadre des processus européens et est reconnue à ce titre.
Ce défi important pour l'Agence, identifié dès la création en 1995 de l'Agence européenne du médicament (EAMA) explique que les autorisations de mise sur le marché (AMM) sont traitées d'une manière et dans des délais satisfaisantes.
La reconnaissance de l'ANMV est illustrée par ses responsabilités européennes :
-- Pour la procédure de reconnaissance mutuelle, la France a été choisie comme Etat-membre de référence dans 40 % des cas pour la période 1995-1998 et 19 % en 2001 et 2002, l'importance prise par l'Irlande expliquant en partie cette diminution.
-- Pour la procédure centralisée, la France a été rapporteur (ou co-rapporteur) dans 30 % des cas pour la période 1995-1998 et 40 % pour 1999-2002.
Les efforts importants consentis dans les procédures européennes semblent avoir pour contrepartie des retards dans procédures d'enregistrement et de traitement des dossiers (pré-AMM, médicaments immunologiques).
Par ailleurs, la pharmacovigilance et le contrôle de la qualité ne sont pas assurés dans des proportions satisfaisantes. Il en est de même pour l'inspection où seule sont suivis les soixante établissements ayant une activité exclusive de production de médicaments vétérinaires.
On doit aussi faire un signalement particulier pour la publicité. Son contrôle (art. R. 146-43 à 47 du code de la santé publique) n'est pas assuré. Cette lacune est d'autant plus préoccupante que les faiblesses et les abus dans le domaine de la prescription et de la distribution des médicaments vétérinaires perdurent dans des proportions inquiétantes et ce, après avoir été clairement mis en évidence par le rapport commun IGAS-COPERCI de mars 2002.
2.7.3. Les raisons des faiblesses
-- L'insuffisance des moyens, notamment en personnel, est l'une des principales raisons des faiblesses de l'ANMV. C'est un élément important dont il a été pris acte à différentes reprises et à tous les niveaux, y compris bien sûr celui des inspections générales. On n'a pas à en donner l'analyse ici, mais il doit évidemment en être tenu compte pour redresser la situation. Or, les tutelles (agriculture et santé) ne portent pas toute l'attention qu'elles devraient alors que pourtant les enjeux sont importants en termes économiques et en termes de santé publique. La crise de l'ESB devrait au moins le rappeler. En outre, les moyens supplémentaires demandés restent en valeur absolue, sinon en pourcentage, d'une modestie qui rend les discussions quelque peu dérisoires (30 emplois permanents et 10 temporaires demandés depuis 2002).
La faiblesse de la synergie que l'on pourrait envisager avec l'intégration dans l'AFSSA a souvent été soulignée. Cela s'illustre de différentes manières. L'une qui mérite plus particulièrement d'être soulignée ici est l'absence d'évolution dans la conduite de l'expertise. En effet, il est souvent constaté, que contrairement à d'autres instances, l'ANMV a une « expertise interne forte et une expertise externe faible ». Cela correspond à une culture d'entreprise où, compte tenu de la spécificité du secteur, l'expert extérieur est considéré avec une certaine méfiance. La composition des commissions d'experts n'a pas évolué, les conditions de recrutement des experts non plus (pas d'appel à candidature ni de comité de sélection).
La localisation de l'ANMV à Fougères est également un élément avancé pour expliquer certaines des difficultés que l'ANMV rencontre dans l'accomplissement de ces tâches ; l'absence de voisinage, d'une école vétérinaire est soulignée dans ce cadre. Le transfert de l'ANMV dans une grande ville universitaire où elle pourrait être « adossée » à une école vétérinaire avec tous les moyens scientifiques et humains que cela signifie, et bénéficier de dessertes rapides nécessaires à l'exercice de ses missions (liens avec Paris et Londres pour l'Agence européenne du médicament) est clairement désigné, comme la condition préalable au succès des mesures de mise à niveau et de réorganisation de certaines fonctions de l'Agence, en particulier pour les différentes facettes de l'expertise, l'inspection et la pharmacovigilance.
Le transfert posera nécessairement des problèmes à l'ensemble des personnels de l'ANMV qui pour beaucoup ont dû consentir un effort personnel pour s'installer à Fougères et seront amenés à en faire d'autres pour un éventuel déménagement. En outre, les conséquences en terme économique et social pour le pays de Fougères doivent être prises et compte et compensées pleinement par un plan d'ensemble, ce qui, à l'échelle des ministères de tutelle concernés, est envisageable.
Il serait plus raisonnable d'améliorer l'intégration de l'ANMV dans le dispositif général de l'AFSSA et de renforcer son articulation avec les centres de recherche vétérinaire, en particulier ceux de l'Ouest.
2.7.4. Une remise en cause de l'intégration dans l'AFSSA ?
L'intégration de l'ensemble du CNEVA à l'AFSSA lors de la création de cette dernière par la loi du 1 er juillet 1998 n'est pas remise en cause aujourd'hui alors que beaucoup doutaient de sa réussite et pour certains de sa faisabilité même. Pour l' ANMV elle-même, la question apparaît sous un jour différent. Lors de l'examen de la proposition de loi sénatoriale, le positionnement institutionnel de l'ANMV fut discuté et l'option d'une intégration à l'AFSSAPS envisagée.
Un tel rattachement, qui est la seule alternative existante à la situation actuelle, s'appuie sur quelques arguments. Le premier est fourni par la comparaison avec la configuration du niveau européen. Le médicament vétérinaire est traité au sein de l'Agence européenne de Londres pour l'ensemble des médicaments (EMEA) et dans la majorité des Etats-membres, les deux catégories de médicaments relèvent dela même structure, étant entendu que la Grande-Bretagne et l'Allemagne ont une entité spécifique au médicament vétérinaire. En second lieu, certaines difficultés que rencontre l'ANMV sembleraient pouvoir être résolues par l'intégration au sein d'une agence dont la priorité des fonctions et de l'objet (le médicament) faciliterait l'approche et le traitement ; en outre, il convient de garder à l'esprit que l'ANMV comme l'AFSSAPS, contrairement à l'AFSSA, ont des compétences qui vont au-delà de l'évaluation du risque puisqu'elles comprennent la gestion du risque.
A l'encontre d'une intégration dans l'AFSSAPS, les arguments sont davantage tirés de l'analyse des situations réelles et des risques potentiels que de considérations juridiques ou liées à l'architecture des structures administratives.
Les spécificités du médicament vétérinaire risqueraient d'être marginalisées, voire négligées, dans un ensemble administratif entièrement dédié à l'application humaine qui doit en outre se consacrer à de nouveaux domaines extérieurs au médicament : dispositifs médicaux, cosmétiques, produits relevant des allégations. On peut concevoir la crainte qu'une certaine culture du médicament vétérinaire se dilue dans l'opération.
Les liaisons de plus en plus d'actualité avec d'autres problèmes de la sphère agricole auraient d'autant moins de chances d'être privilégiées : résidus, l'écotoxicité alors même que les questions des produits phytosanitaires, engrais, biocides, doivent faire l'objet, par ailleurs, de réformes nécessaires.
Sur le plan structurel, le positionnement de la tutelle du ministère de l'agriculture deviendrait problématique, une tutelle partagée sur l'AFSSAPS étant inenvisageable. Enfin, le choc que constituerait le transfert géographique de l'ANMV couplé à un changement de structure de ce type rendrait sans doute l'ensemble de l'opération par trop délicate. Au total, malgré les difficultés et insuffisances constatées, la pertinence de l'appartenance à l'AFSSA se vérifie.
La confirmation de l'intégration au sein de l'AFSSA ne devrait donc pas faire de doute. C'est plutôt l'effectivité de celle-ci qui devrait être renforcée, les progrès à réaliser se situant aussi du côté de la direction générale de l'AFSSA, la question des moyens, quant à elle, interpellant directement les tutelles.
III. LA SÉCURITÉ SANITAIRE DES ALIMENTS ET L'EUROPE
3.1. Deux mots d'histoire récente
La crise de la « vache folle » (ESB) a bouleversé l'approche des questions de sécurité sanitaire des aliments en Europe au cours des années quatre-vingt-dix, il y a maintenant plus de dix ans. Outre l'ampleur de la crise qui a pris naissance en Grande-Bretagne, l'extension au niveau européen n'a été ni envisagée ni gérée à la mesure du problème et des risques par les instances communautaires compétentes. Les effets particulièrement dommageables et durables pour l'ensemble de ces instances a heureusement, mais tardivement, provoqué un réaménagement substantiel du dispositif sanitaire. Les notions de libre circulation des marchandises et de marché unique, sans être remises en cause, ont dû laisser un espace aux considérations de santé des consommateurs. C'est ainsi qu'a été créée en 1997 au sein des services de la Commission européenne la Direction générale Santé et protection des consommateurs.
Préalablement en 1992, une directive a prévu, pour la première fois un échange obligatoire de données entre Etats-membres sur les agents responsables des toxi-infections alimentaires. Mais la disparité des données recueillies a été à l'origine de grandes difficultés pour atteindre un niveau opérationnel.
En 1993, la directive 93/43 relative à l'hygiène des services alimentaires a constitué un pas supplémentaire imposant un corpus de règles visant les différentes étapes de la production. Néanmoins, la nécessité d'une meilleure lisibilité et d'une homogénéisation des procédures et des dispositions subsistait. C'est dans ce contexte que le livre blanc de la Commission a appréhendé en 2000 l'ensemble du problème.
Les priorités de la Commission sont alors les suivantes (28 ( * )) :
- Contribuer à la meilleure protection possible des consommateurs européens en mettant en oeuvre une politique qui intègre l'ensemble des préoccupations des filières alimentaires, de la ferme à la fourchette ;
- Regagner la confiance des consommateurs dans la capacité des industries alimentaires et des pouvoirs publics à garantir la sécurité des aliments ;
- Définir les bases juridiques et techniques permettant de garantir cette sécurité en prenant notamment en compte les problèmes sanitaires liés à l'alimentation;
- Se doter d'un nouveau dispositif législatif et d'une agence européenne de sécurité alimentaire.
C'est donc à l'issue de ce long et difficile processus qu'a été mise en place l'Autorité Européenne de Sécurité des aliments à l'automne 2002.
Les nouvelles législation et réglementation en préparation depuis longtemps ont été acquises récemment le 26 avril 2004 avec l'adoption de ce que l'on appelle le « paquet hygiène » sur la base du règlement 178/2002 du Parlement Européen et du Conseil en date du 28 janvier 2002. Celui-ci établit « les principes généraux et les prescriptions générales de la législation alimentaire instituant l'Autorité européenne de sécurité des aliments et fixant des procédures relatives à la sécurité des denrées alimentaires ». L'entrée en vigueur de ces textes, qui ont été adoptés à l'issue d'une procédure de co-décision avec le Parlement Européen, est fixée au 1 er janvier 2006.
Après une très rapide indication de ce qu'est le « paquet hygiène », on abordera la mise en place de l'Autorité Européenne de Sécurité des Aliments, puis on caractérisera la nouvelle situation d'ensemble avec la clarification qu'elle appelle. On traitera plus particulièrement un élément essentiel dans la nouvelle architecture communautaire : le réseau des instances compétentes en matière de sécurité des aliments.
3.2. Le « paquet hygiène »
Les quatre volets de ce nouvel ensemble de règles qui se substituent à tout ce qui préexistait dans ce domaine comprend :
-- les règles générales d'hygiène applicables à toutes les denrées alimentaires ;
-- les règles spécifiques d'hygiène applicables aux denrées alimentaires d'origine animale :
-- les règles spécifiques d'organisation des contrôles officiels applicables à ces dernières denrées (viandes, produits de la pêche, coquillages, lait et produits laitiers). Ces règles viennent en complément de celles relatives aux aliments pour animaux : en avril 2003, la Commission avait adopté un règlement fixant de nouvelles exigences dans ce domaine (additifs par exemple), mais certains aspects n'étaient pas réglementés ainsi la production, le transport, le stockage et la manipulation des aliments pour animaux.
Sans s'attarder sur un sujet qui reste extérieur au présent rapport, mais qui éclaire un peu l'environnement institutionnel, signalons que certains interlocuteurs ont fait part d'interrogations, voire d'inquiétudes, sur la mise en application de cet ensemble de nouvelles dispositions communautaires, indiquant notamment que l'acception de ces textes variait beaucoup d'un acteur à un autre.
Pour ce qui nous concerne, nous nous contenterons de relever les perspectives d'évolution des fonctions de contrôle, notamment telles qu'elles sont indiquées dans le document communautaire NEMO/04/95 du 26 avril 2004 sous forme de questions réponses 29 ( * ) :
« Règles relatives aux inspections et aux contrôles
Qui est responsable de l'inspection des viandes ?
L'inspection des animaux vivants et des animaux morts s'effectue sous la responsabilité du vétérinaire officiel qui peut être assisté par des auxiliaires officiels et, dans des circonstances déterminées et uniquement pour la viande de volaille et de lapin, par le personnel de l'abattoir. Une formation complète et détaillée est requise pour les vétérinaires, les auxiliaires et le personnel des abattoirs qui participent à l'inspection des viandes. Au moins un vétérinaire officiel doit être présent dans l'abattoir tout au long des inspections des animaux vivants et des animaux morts (les inspections dites ante mortem et post mortem). Il en va de même lors de l'inspection post mortem dans les établissements de traitement du gibier.
L'inspection des animaux vivants est-elle possible dans l'exploitation ?
L'inspection dans l'exploitation par un vétérinaire agréé est possible. Pour les porcins par exemple, les animaux doivent être accompagnés d'un certificat sanitaire et doivent être abattus dans les trois jours qui suivent l'inspection.
En quoi les nouvelles règles modifieront-elles l'inspection des viandes ?
Les nouvelles règles autorisent une approche plus moderne fondée sur l'évaluation des risques, ce qui signifie que, dans certaines conditions, il est possible de limiter l'inspection post mortem à un examen visuel sauf, bien entendu, lorsque des anomalies sont détectées. Une telle procédure d'inspection pourrait, par exemple, s'appliquer à des porcs d'engraissement maintenus dans des conditions de logement contrôlées dans des systèmes de production intégrés.
A l'avenir, les tâches traditionnelles d'inspection des viandes du vétérinaire officiel seront progressivement remplacées par des tâches d'audit. Ainsi, le vétérinaire officiel devra vérifier la mise en oeuvre du système HACCP, ce qui comportera une évaluation des points de contrôle critiques, le contrôle des registres journaliers, la vérification de la bonne application des procédures d'hygiène, etc. L'inspection des viandes ne sera toutefois pas privatisée. Le vétérinaire officiel est responsable en dernier ressort de l'inspection des viandes, même s'il peut être assisté par des auxiliaires dûment formés à cet effet. Dans les établissements qui abattent de la volaille et des lagomorphes (des lapins par exemple), le vétérinaire peut aussi, dans certaines conditions, être assisté par les personnels de l'abattoir. Ces derniers n'ont toutefois pas exactement les mêmes fonctions que les auxiliaires et ne peuvent pas, par exemple, accomplir des tâches d'audit.
(...)
Quel est l'impact des règles d'hygiène sur les importations de denrées alimentaires d'origine animale ?
Les produits d'origine animale importés doivent respecter les normes strictes de l'UE en matière de sécurité des aliments, y compris les règles d'hygiène. L'importation de ces produits est dès lors uniquement autorisée à partir de pays et d'établissements qui figurent sur une liste communautaire gérée par la Commission européenne pour le compte des Etats membres».
L'évolution de l'activité des services vétérinaires vers des tâches d'audit est une orientation qui a été prise pour tenir compte de l'évolution observée dans plusieurs Etats-membres où certaines tâches de contrôle sont déjà délégués, d'après les commentaires autorisés faits sur ces textes.
Ces perspectives, fondées apparemment sur l'objectif d'aligner les règles sur la pratique doivent pour le moins être explicitées et sérieusement encadrées afin d'éviter qu'elles ne soulèvent l'inquiétude. Cet éclairage est pertinent : à une question sur le volume global des activités de l'Office alimentaire et vétérinaire (OAV) de la Commission, il a été précisé que celui-ci « ne diminuera pas ; néanmoins, le nouveau système d'audits généraux permettra une approche plus intégrée des inspections et une meilleure utilisation des ressources disponibles ».
On aurait pu s'attendre, compte tenu de l'extension du domaine de compétences et des fonctions, à ce que ce volume soit appelé à croître sensiblement en raison de l'entrée de dix nouveaux Etats-membres. Dans ce contexte, les moyens de l'Office communautaire à Dublin paraissent pour le moins avoir besoin d'une substantielle mise à niveau et ses conditions de fonctionnement doivent être nettement améliorées.
Enfin, constituant cette fois-ci un élément important dans le renforcement de la sécurité qui mérite d'être signalé, le règlement européen 178/2002 (chapitre 18) est entré en vigueur le 1 er janvier 2005. Il impose la traçabilité des produits alimentaires à toutes les étapes de la production et de la distribution afin que le rappel des produits puisse s'effectuer en toute circonstance. Ce progrès, qui comporte certes des coûts non négligeables pour l'ensemble des filières agro-alimentaires, répond à une nécessité que les crises ont mis en lumière.
3.3. La mise en place de l'AESA (Autorité européenne de sécurité des aliments)
L'AESA qui constituait une proposition essentielle du livre blanc a été instituée en janvier 2002 par le règlement n° 178/2002 du Parlement Européen et du Conseil, mais sa mise en place a, logiquement, exigé un délai nécessité, notamment par les nominations des différents responsables et instances :
-- le conseil d'administration en septembre 2002,
-- le directeur exécutif, M. Geoffrey Podger, britannique issu de la Food Standards Agency, en octobre 2002 qui a pris ses fonctions au début de l'année 2003.
C'est en mai 2003 que l'Autorité a pu démarrer ses activités. La fixation de la composition du conseil d'administration (14 membres) ne s'est pas faite sans difficulté et qu'elle a pris presque un an. Tous les Etats-membres d'alors y sont représentés à l'exception du Luxembourg. Le Britannique qui y siège est le (seul) représentant des associations de consommateurs. La Commission, quant à elle, dispose d'un siège qui s'ajoute aux 14 qui se recrutent « de manière à assurer le niveau de compétence le plus élevé, une expertise diversifiée ainsi que la plus large représentation géographique ».
Les tâches de l'Autorité ont ainsi été précisées par l'art. 23 du Règlement 178/2002 :
« a) fournir aux institutions de la Communauté et aux Etats-membres les meilleurs avis scientifiques possibles dans tous les cas prévus par la législation communautaire ainsi que sur toute question qui relève de sa mission ;
b) promouvoir et coordonner la mise au point de méthodes uniformes d'évaluation des risques dans les domaines relevant de sa mission ;
c) fournir une assistance scientifique et technique à la Commission dans les domaines relevant de sa mission et, lorsqu'elle en fait la demande, pour l'interprétation et l'examen des avis sur l'évaluation des risques ;
d) commander les études scientifiques nécessaires à l'accomplissement de sa mission ;
e) rechercher, recueillir, rassembler, analyser et résumer les données scientifiques et techniques dans les domaines qui relèvent de sa mission ;
f) mener une action d'identification et de caractérisation des risques émergents, dans les domaines qui relèvent de sa mission ;
g) établir un système de réseaux des organismes opérant dans les domaines qui relèvent de sa mission et en assurer le fonctionnement ;
h) fournir une assistance scientifique et technique, lorsque la demande lui en est faite par la Commission, dans le cadre des procédures de gestion des risques mises en oeuvre par la Commission en matière de sécurité des denrées alimentaires et des aliments pour animaux ;
i) apporter, lorsque la demande lui en est faite par la Commission, un soutien scientifique et technique en vue d'améliorer la coopération entre la Communauté, les pays ayant introduit une demande d'adhésion, les organisations internationales et les pays tiers, dans les domaines qui relèvent de sa mission ;
j) veiller à ce que le public et les parties intéressées reçoivent rapidement une information fiable, objective et compréhensible dans les domaines qui relèvent de sa mission ;
k) exprimer de manière autonome ses propres conclusions et orientations sur les questions qui relèvent de sa mission ;
l) effectuer toute tâche qui lui est assignée par la Commission dans le domaine qui relève de sa mission ».
Il apparaît que les compétences de l'AESA sont déterminées de façon tout à fait comparable à celles de l'AFSSA, compte tenu des différences structurelles et fonctionnelles de principe entre une agence nationale et une agence communautaire. L'AESA a la compétence complète en matière d'évaluation du risque (y compris sur les OGM où pour les raisons historiques que l'on a signalées, l'AFSSA n'a qu'une compétence partielle) et la gestion du risque est du ressort de la Commission européenne. La préexistence de l'AFSSA n'y est sans doute pas étrangère et les législateurs français de 1998 souhaitaient d'ailleurs que cette antériorité puisse servir de référence à l'instance européenne à créer.
Cette similitude de situations doit d'autant plus être remarquée que cette configuration des compétences est nettement minoritaire parmi les Etats-membres qui ne disposent pas tous d'une agence autonome dans le domaine de la sécurité des aliments. Le directeur exécutif de l'AESA a fait part d'une perception quelque peu différente des situations, estimant qu'il y avait une partie de la gestion du risque qui en France relevait de l'AFSSA.
Le décalage dans l'appréciation des réalités a peut être pour origine « l'évaluation de la gestion du risque », notamment par la transmission des plans de contrôles et des réalisations qui incombe à la DGAL ET la DGCCRF principalement. Par ailleurs, l'AESA ne dispose pas du tout de laboratoire et en outre, elle n'a pas de compétence dans le domaine du médicament vétérinaire qui est du ressort de l'Agence européenne du médicament de Londres.
L'AESA, et c'est aussi une différence qui peut expliquer ce décalage, n'a pas davantage de compétence en matière de recherche, contrairement à l'AFSSA. A travers la Direction générale recherche, la Commission dispose, elle, d'importants moyens, notamment d'orientation. Parmi les 13 directions que compte les D.G., l'existence de la direction « Sécurité des systèmes de production alimentaire » a montré l'intérêt apporté à ces questions ainsi que les moyens mobilisés avec 685 millions d'euros pour le domaine thématique prioritaire « qualité et sécurité alimentaire » au titre du 6° PCRD (2002-2005). Parmi les 12 programmes que comporte ce domaine, l'AFSSA a été retenue comme coordinateur du programme Medvetnet (réseau médecine vétérinaire) sur les zoonoses. C'est là une illustration de la valeur de l'activité de l'AFSSA dans le domaine de la recherche.
Si la mise en place institutionnelle de l'AESA est maintenant acquise, depuis qu'elle a commencé à rendre des avis il y a un peu moins d'un an, son installation matérielle et fonctionnelle est loin d'être achevée. Matériellement, l'AESA compte un peu moins d'une centaine d'agents ; elle devrait atteindre un effectif de 250 à 300 une fois complètement déployée dans deux à trois ans. Mais ce déploiement passe d'abord par l'installation, donc le déménagement depuis Bruxelles jusqu'à Parme en Italie. De préférence à Helsinki, et plus lointainement Lille et Barcelone, Parme a en effet été choisie en décembre 2003 comme siège définitif, en même temps qu'étaient effectués les choix des sièges d'une douzaine d'agences européennes.
La mise en place fonctionnelle sera plus avancée lorsque, le mouvement se prouvant en marchant, les mécanismes d'évaluation auront suffisamment fonctionné avec des résultats tangibles dans un domaine où les autres acteurs étant nombreux et attentifs, l'évaluation du risque sera courante et réalisée si possible dans un paysage européen coordonnée où les rôles de chacun seront utilement articulés. C'est là la plus grande question qui se pose à la fois pour l'AESA et pour l'AFSSA elle-même.
3.4. Une clarification nécessaire
Les compétences de l'AESA sont déterminées en termes clairs. Elles cadrent son fonctionnement et la positionnent face aux autres acteurs du domaine. On pense, en particulier, à la Commission qui est, notamment, gestionnaire du risque et coordonne la recherche. Des difficultés limitées et ponctuelles peuvent d'ailleurs apparaître dans les relations entre l'AESA et la Commission ; l'avenir dira si elles sont liées à la phase transitoire d'installation, à des questions relationnelles entre responsables ou plus durablement à d'autres facteurs.
Le positionnement de l'AESA avec les agences ou instances nationales des Etats-membres constitue un problème d'évidence plus important et exigeant beaucoup de soin de part et d'autre si l'on veut que l'ensemble du mécanisme de la sécurité sanitaire des aliments en Europe fonctionne sans difficulté majeure.
Au début de son fonctionnement, l'Autorité ne peut traiter à elle seule tous les dossiers dont la Commission, les Etats-membres, le Parlement européen pourraient la saisir, sans oublier son pouvoir d'auto-saisine. Mais il est possible qu'à terme elle envisage de jouer dans tous les domaines un rôle central, laissant aux agences nationales un rôle de sous-agences, d'auxiliaire compétent principalement pour des questions locales et la communication adaptée à chaque Etat-membre. Cette perspective apparaît pour l'instant peu probable car les dispositions du Règlement cadrent nettement le mécanisme sur la mutualisation des activités et le fonctionnement en réseau, sans parler du principe de subsidiarité.
C'est donc dans le sens d'une coopération entre l'AESA et les agences nationales sur la base de ces principes que l'on peut espérer s'orienter. Cela veut dire notamment des mécanismes d'échanges d'informations, un partage des tâches coordonné pour une part importante des travaux d'évaluation et une information opérationnelle des agences nationales avant la publication des avis ; par « information opérationnelle », on entend permettant une réaction dans un délai suffisant.
La nécessité d'une bonne articulation entre l'Autorité européenne et les agences nationales a été bien identifiée dans le règlement de 2002 et fait l'objet de nombreuses dispositions à cette fin.
Le Forum consultatif (art. 27) est un élément essentiel dans cet ensemble : on développera ce point avant la conclusion du présent développement. Il en sera de même du « réseau d'organismes opérant dans les domaines qui relèvent de la mission de l'Autorité », tel qu'il est prévu à l'art. 36.
Dans ce qui est le « coeur de métier » de l'AESA, c'est-à-dire la production d'avis scientifiques sur l'évaluation des risques régi par l'art. 29, il est prévu spécifiquement à l'art. 30 la situation et les procédures en cas « d'avis scientifiques divergents ».
Des divergences de ce type ont été enregistrées dans deux dossiers récents : celui des OGM et celui du semicarbazide. Dans ce dernier cas (il s'agit d'un « matériau-contact » utilisé notamment dans les pots d'aliments pour bébés), il y eu des divergences d'appréciation du risque entre l'AESA d'une part et l'AFSSA, de même que la « Food Standards Agency » britannique ; en outre, la Commission européenne a eu tendance à considérer que l'AESA débordait, par ses recommandations, sur le rôle de gestionnaire du risque qui est le sien.
S'agissant d'un OGM, le maïs doux « Bt11 » destiné à l'alimentation humaine, l'avis favorable au niveau européen était celui du comité scientifique sur l'alimentation humaine rendu en avril 2002, antérieurement à la mise en place de l'AESA, mais il a été renouvelé le 19 avril 2004 ; l'AFSSA de son côté a émis successivement deux avis exigeant de nouvelles études pour autoriser la consommation de maïs Bt11, dont le dernier le 15 avril 2004, avis dont la conclusion est la suivante :
« L'Agence française de sécurité sanitaire des aliments maintient son avis du 26 novembre 2003 qui concluait qu'en toute rigueur, pour évaluer l'impact d'une consommation régulière de maïs doux portant l'événement de transformation Bt11, il conviendrait de disposer d'une étude de toxicité/tolérance chez le rat ou d'une étude de tolérance/alimentarité chez un animal d'élevage (par exemple, le poulet de croissance) réalisée avec du maïs doux portant cet événement de transformation.
« L'Agence française de sécurité sanitaire des aliments est consciente qu'une telle étude n'est pas exigible dans le cadre de la réglementation actuelle, mais qu'elle pourrait être souhaitable du fait que le métabolisme et la physiologie du maïs doux diffèrent sensiblement de ceux du maïs « champ » et que le maïs doux est le seul destiné à être consommé en l'état par l'homme ».
Il est à noter que c'est ce maïs dont la vente (maïs importé) a été autorisée le 19 mai 2004 par la Commission européenne (faute d'accord au niveau du Conseil européen le 26 avril 2004). Cette décision était conditionnée à l'entrée en vigueur (le 18 avril 2004) des dispositions relatives à la traçabilité.
Sur des saisines de l'AESA, l'AFSSA a rendu des avis ou des rapports de manière partielle. L'Agence Française, interrogée par votre rapporteur, en a indiqué pour le dernier trimestre 2003 sept à titre d'exemples : l'une des saisines (évaluation des risques des acides gras trans dans les aliments) coïncide exactement pour les deux agences ; il a dans cette perspective été précisé par l'AFSSA : « un groupe de travail est d'ores et déjà activé à l'AFSSA sur ce sujet avec la perspective d'un rapport dans six mois. Il conviendra donc sur ce sujet d'optimiser les échanges avec l'AESA afin d'éviter une duplication du travail d'évaluation ».
Sur les sujets d'études en cours dans les deux agences, la réponse de l'AFSSA est la suivante :
« Le programme de travail de l'AESA récemment communiqué fait apparaître peu de sujets d'étude commun sur des thématiques générales.
Il peut être noté parmi les sujets d'étude de l'AESA :
-- nitrates/nitrites dans les viandes sous l'angle microbiologique (tout récemment achevé) ; L'AFSSA est saisie sur des aspects plus vastes de la problématique des nitrates (incluant l'eau) ;
-- approche de l'allergénicité des OGM ; l'AFSSA envisage une réflexion également sur ce thème au sein de son CES compte tenu des diverses activités développées dans ce domaine ;
-- mercure dans les aliments ; l'AFSSA poursuit sa réflexion sur le méthyl mercure dans les poissons (un avis déjà publié) ;
-- contaminants PCB non dioxines like ; l'AFSSA poursuit sa réflexion sur le sujet (plusieurs avis déjà publiés) ;
-- Ochratoxine A ; l'AFSSA initie une réflexion générale sur les mycotoxines dans la chaîne alimentaire de l'homme et de l'animal ;
-- Semicarbazide et acrylamide ; ces sujets se poursuivent à la fois au niveau communautaire et à l'AFSSA (projets de recherche, acquisition de données de contamination).
Sur l'ensemble de ces sujets difficiles, une information réciproque doit être mise en place dans le cadre du réseau européen ».
« Sur les ESST, de nombreux sujets d'étude sont envisagés au niveau européen et par l'AFSSA. D'ores et déjà, l'Autorité européenne de sécurité des aliments (AESA) a transmis à l'AFSSA deux saisines dont elle a fait l'objet de la part de la Commission européenne.
Ces thématiques font ou ont fait l'objet d'une évaluation de l'AFSSA, qui pourra apporter certaines de ses conclusions ou de ses éléments de réflexion à l'Autorité européenne ».
Des évaluations récentes laissent donc à penser que la coopération est souvent réelle, même pour les ESST où pourtant il y a un passé difficile.
Au-delà des difficultés évidentes que pourraient soulever la survenance d'avis scientifiques divergents, mais dont on voit qu'il ne faut pas exagérer la probabilité, c'est l'ensemble de la cohérence du mécanisme qui doit être recherché car le besoin est avéré et certaines évolutions pourraient lui conférer une réelle importance.
-- Les groupes scientifiques spécialisés constituent l'élément essentiel du dispositif d'évaluation du risque. Ils préexistaient à l'AESA et ceux qui existaient lors de la mise en place de celle-ci avaient été formés en 1997. Cinq d'entre eux ont été transférés à l'AESA qui, avec les nouvelles créations, en compte désormais huit outre le comité scientifique (multi sectoriel). Les groupes qui ont donc une ancienneté nettement supérieure à l'AESA elle-même ne semblent pas poser de problème particulier dans leur fonctionnement. En revanche, leur positionnement par rapport à ceux des agences nationales pourrait susciter des problèmes dès lors que des doublons apparaîtraient sur les sujets de saisine ou thème d'études. En cas de double appartenance, la charge de travail et les contraintes pour les experts concernés pourraient être source de difficultés.
Le recrutement des experts lui-même, opération lourde et délicate, exige une certaine coordination entre les instances compétentes européennes et nationales. Si il n'est pas pensable que l'AESA doive passer par les agences nationales pour recruter des experts même pour des sujets spécifiques en vue d'une étude ponctuelle, il n'est pas davantage concevable qu'il n'y ait pas coordination et que, d'une manière générale il n'y ait pas une recherche coutumière d'harmonisation des procédures et des calendriers, sans parler des consultations informelles.
L'insuffisante considération reconnue aux experts dans les milieux universitaires, et pas seulement en France, peut être notamment contrebattue par une harmonisation par le haut des conditions de l'expertise en Europe. L'AESA aura sans doute les moyens de participer à une telle évolution nécessaire. Il ne serait pas souhaitable, en revanche, qu'une concurrence mal coordonnée entraîne un moindre intérêt pour l'expertise au niveau national. L'inverse n'est pas à exclure si le recrutement des experts au niveau européen prenait davantage en compte des critères de représentativité géographique que l'excellence professionnelle. On remarque ainsi que la coordination et la mutualisation prévue dans le Règlement de janvier 2002, notamment son article 36 correspond à une vraie nécessité.
Une concrétisation des mécanismes de coopération
-- « Le forum consultatif (art. 27 du règlement) se compose des représentants des Etats-membres qui accomplissent des tâches analogues à celles de l'AESA à raison d'un par Etat-membre ». Le forum conseille le directeur exécutif, notamment en vue de l'élaboration d'une proposition relative au programme de travail de l'Autorité. Le directeur exécutif peut également demander l'avis du forum sur la hiérarchisation des demandes d'avis scientifiques.
Cet article précise notamment :
« Le forum consultatif constitue un mécanisme pour l'échange d'informations sur les risques potentiels et la mise en commun des connaissances. Il veille au maintien d'une étroite coopération entre l'Autorité et les instances compétentes des Etats-membres, en particulier :
Pour éviter tout double emploi des études scientifiques de l'Autorité avec les programmes des Etats-membres, conformément à l'article 32 ;
Dans les cas visés à l'article 30, paragraphe 4, lorsque l'Autorité et un organisme national sont tenus de collaborer ;
Pour promouvoir le fonctionnement en réseaux européen des organismes opérant dans les domaines qui relèvent de la mission de l'Autorité, conformément à l'article 36, paragraphe 1 ;
Lorsque l'Autorité ou un Etat-membre identifie un risque émergent.
Le forum consultatif est présidé par le directeur exécutif. Il se réunit régulièrement à l'invitation du président ou à la demande d'au moins un tiers de ses membres, au moins quatre fois par an ».
Le forum se réunit effectivement quatre fois par an actuellement et semble fonctionner dans des conditions qui s'inscrivent dans la perspective d'une philosophie de la coopération entre instances nationales, ce qui n'empêche pas celles-ci de se réunir éventuellement entre elles sur des sujets généraux, d'ordre nutritionnel par exemple.
Le fonctionnement en réseau est prévu d'une manière très claire par l'article 36 du Règlement :
« Article 36 : Réseau d'organismes opérant dans les domaines qui relèvent de la mission de l'Autorité.
L'Autorité favorise le fonctionnement en réseaux européens des organismes opérant dans les domaines qui relèvent de sa mission. Ce fonctionnement en réseaux a pour objectif, en particulier, de promouvoir un cadre de coopération scientifique en facilitant la coordination de l'action, l'échange d'informations, l'établissement et l'exécution de projets communs,l'échange de connaissances spécialisées et de meilleures pratiques dans les domaines qui relèvent de a mission de l'Autorité.
Le Conseil d'administration, sur proposition du directeur exécutif, établit une liste rendue publique des organismes compétents désignés par les Etats-membres qui, soit individuellement, soit dans le cadre d'un réseau, peuvent aider l'Autorité dans sa mission. L'Autorité peut confier à ces organismes certaines tâches, en particulier des travaux préparatoires aux avis scientifiques, des tâches d'assistance scientifique et technique, la collecte de données et l'identification des risques émergents. Certaines de ces tâches peuvent bénéficier d'un soutien financier (...) ».
L'articulation de la coopération bien décrite ici tarderait à se concrétiser. A ce titre, des impulsions en provenance des Etats-membres eux-mêmes seraient de nature à développer les mécanismes de coopération en faisant concrètement avancer le partage des tâches pour une réelle efficacité évitant ainsi les risques de dérives ultérieures. A titre d'exemple, en ce qui concerne les « risques émergents » des procédures adaptées impliquant à l'avance tous les acteurs permettraient un réel progrès.
Les progrès réalisés par la mise en place de ces mécanismes devaient être constatés dans un inventaire dans les termes suivants (art. 36, alinéa 4).
« Dans un délai d'un an à compter de l'entrée en vigueur du présent règlement, la Commission publie un inventaire des systèmes communautaires existant dans les domaines qui relèvent de la mission de l'Autorité et prévoyant que les Etats-membres effectuent certaines tâches d'évaluation scientifique, notamment dans le cadre de l'examen des dossiers d'autorisation. Ce rapport, accompagné le cas échéant de propositions, indique notamment pour chaque système existant, les modifications ou améliorations qui sont éventuellement nécessaires pour permettre à l'Autorité d'accomplir sa mission, en coopération avec les Etats-membres ».
Il serait heureux que, plus d'un an après la mise en place effective de l'AESA, on ait prochainement le moyen de mesurer ainsi le fonctionnement en réseau de celle-ci.
*****
**
Pour conclure, on ne peut que souligner les résultats très positifs du travail et de l'action de l'AESA, de l'AFSSA, de la Food Standards Agency et des autres acteurs lors du trouble connu par la filière saumon l'hiver dernier (janvier 2004). Un article de revue dont le caractère scientifique douteux a pu être rapidement et avec certitude mis en cause a illustré l'absence de fondement des inquiétudes soulevées. Certes, les ventes de saumon ont connu une baisse sensible pendant quelques semaines, mais en d'autres temps, on aurait sans doute vu se développer une peur que l'on aurait très abusivement qualifiée de « crise sanitaire ». C'est là un signe de progrès que l'on relève avec intérêt.
RESUME DE LA DEUXIEME PARTIE
|
Agence entièrement nouvelle créée en 1998, l'AFSSA a répondu aux attentes et a atteint les objectifs qui lui étaient assignés ; des ajustements restent à réaliser et surtout certains rapports doivent être mieux définis avec les autres acteurs de la sécurité des aliments, notamment au niveau européen. Une évaluation globalement positive Chargée de l'évaluation du risque pour l'ensemble du domaine de la sécurité sanitaire des aliments, l'AFSSA a su rapidement monter en puissance alors que le climat de crise perdurait avec les rebondissements relatifs à l'ESB (embargo sur la viande britannique, matériaux à risque, abattage sélectif) : la construction ex-nihilo du coeur de l'appareil d'évaluation qu'est la DERNS (Direction de l'évaluation des risques nutritionnels et sanitaires) en est l'illustration la plus concrète. L'intégration réussie des laboratoires du CNEVA (Centre national d'études vétérinaires et alimentaires) est également à souligner. Elle a permis à l'AFSSA d'atteindre la masse critique pour l'expertise, pour l'appui scientifique et technique ainsi que la recherche. La crédibilité de l'Agence dans son ensemble s'est rapidement établie, notamment en tant qu'autorité indépendante d'évaluation des risques ; cette constatation est le fait tant des connaisseurs les plus expérimentées du domaine que de ceux, extérieurs aux circuits administratifs, qui ont à en connaître par leurs fonctions professionnelles ou associatives, ainsi que des observateurs étrangers. Dans le cadre des compétences fort larges qui sont les siennes, les avis de l'AFSSA sont attendus, appréciés et rendus dans des conditions satisfaisantes même si certaines réserves peuvent être formulées sur les modalités de la communication de l'Agence. Le positionnement nutritionnel de l'AFSSA, que la loi lui a reconnu, est essentiel et elle fait face ici aussi à des responsabilités qui sont devenues particulièrement lourdes avec le problème considérable, et en aggravation accélérée, que constitue l'obésité. Au-delà de la réussite, quelques ajustements -- Le principe de séparation entre l'évaluation et la gestion du risque est largement validé par l'expérience. Les quelques remises en cause qui ont pu se faire jour ne justifient pas la réouverture d'un quelconque débat. En revanche, des progrès restent à faire pour que ce principe soit pleinement appliqué ; il faut en somme que dans ce concert de la sécurité sanitaire des aliments où interviennent plusieurs acteurs majeurs, chacun joue le jeu loyalement et donc efficacement. Concrètement, cela implique que l'Agence ait un mode de communication de ses avis qui laisse une marge au gestionnaire du risque (décideur ministériel). Cela exige aussi que les tutelles des trois directions générales sachent précisément exercer cette fonction de tutelle en respectant les principes de la nouvelle architecture administrative définie en 1998. Cela veut dire enfin que dans les tâches pour lesquelles il existe des interfaces, le partenaire administratif chargé de la gestion du risque n'ignore pas ses obligations (plans de contrôle avec la DGAL par exemple). -- Les saisines de l'Agence constituent un mécanisme perfectible. En effet, malgré un fonctionnement actuellement satisfaisant, l'analyse montre des dérives de divers ordres. Le volume de l'ensemble des saisines apparaît à la limite supérieure, la justification de certaines n'est pas avérée, en particulier les saisines automatiques sur tous les projets de textes administratifs. L'instauration d'un mécanisme de sélection s'impose et la formation restreinte du conseil scientifique pourrait notamment être utilisée à cette fin. L'exercice de l'auto-saisine paraît devoir de son côté être mieux cadré. Enfin, le droit de saisine par les industriels, actuellement inexistant, pourrait être reconnu, mais dans un encadrement strict lui aussi. -- L'expertise tout en étant satisfaisante, peut soulever des questions, qui ne sont pas spécifiques au domaine alimentaire (nombre, statut, rémunération des experts). -- La lacune identifiée avec certitude est celle du non respect de la loi de 1998 sur les produits phytosanitaires : clairement inclus dans le périmètre de l'AFSSA, ils restent exclus au niveau de l'application. La situation doit être redressée, les péripéties survenues avec le « Gaucho » et le « Régent » illustrent encore davantage cette nécessité. Le partage excessif des procédures d'expertises relatives aux OGM doit lui aussi être corrigé. -- Quelques aspects des activités de l'AFSSA font l'objet de débats récurrents : place de la recherche, structuration des laboratoires ; les orientations prises depuis 1998 sont positives et l'ancrage de l'Agence nationale du médicament vétérinaire au sein de l'AFSSA doit être confirmé. La sécurité sanitaire des aliments et l'Europe -- Après les redressements indispensables que la crise de l'ESB exigeait, le niveau de sécurité atteint en Europe est élevé, mais incertain. L'insuffisance des contrôles aux frontières de l'Union, les effets non maîtrisés de l'adhésion de dix nouveaux membres sont des facteurs d'inquiétude qui sont d'ailleurs reconnus. Certains principes d'organisation contenus dans le « paquet hygiène » adopté l'an dernier posent des interrogations, même si par ailleurs la traçabilité fait de réels progrès avec le règlement entré en vigueur au 1 er janvier 2005. -- Enfin, le positionnement de l'Agence européenne de sécurité des aliments (AESA) dans l'ensemble du réseau des agences nationales fait problème. Le respect de l'esprit et des textes qui président à cette organisation s'impose, ce qui ne semble pas encore acquis dans toutes les dimensions de l'activité de l'Agence. |
TROISIÈME
PARTIE :
L'AFSSAPS ET LA SÉCURITÉ DES PRODUITS DE
SANTÉ
Le positionnement de l'évaluation
L'AFSSAPS est parmi les agences et les entités autonomes créées par les loi du 1 er juillet 1998 et du 9 mai 2001, celle qui a été soumise au plus grand nombre d'examens par les instances administratives et parlementaires.
En effet, la Cour des Comptes a établi un relevé de constatations définitives le 25 février 2003 qui portait essentiellement sur les conditions de mise en place de l'Agence ; elle a également abordé « les conditions de fonctionnement des agences sanitaires » dans le chapitre X de son rapport sur l'application des lois de financement de la sécurité sociale en septembre 2002.
De leur côté, à la demande de la Direction générale de la santé, un audit de l'AFSSAPS a été établi conjointement en décembre 2002 par l'inspection générale des finances et l'inspection générale des affaires sociales ; les sept inspecteurs en charge de cet audit ont détaillé leurs travaux à travers douze rapports particuliers et un rapport de synthèse.
Enfin, la commission des finances du Sénat a chargé notre collègue, M. Adrien Gouteyron, d'un rapport d'information sur l'AFSSAPS qui a été adopté par cette commission le 16 juillet 2003. Les informations abondantes ainsi réunies récemment sont non seulement précises, mais encore permettent d'étayer une évaluation à l'aide de travaux que seules des instances juridictionnelles et d'inspection ont les moyens de mener.
Dans ces conditions, l'analyse de l'AFSSAPS à laquelle il est procédé ici se situe dans la perspective de l'évaluation telle qu'elle est prescrite par la loi du 1 er juillet 1998 et des enseignements que l'on peut concrètement tirer. Toutefois, les développements intervenus récemment dans le domaine du médicament et plus particulièrement dans celui de l'expertise obligent à envisager comment les structures actuelles peuvent faire face à ces nouveaux défis.
Deux observations préalables doivent être faites pour éclairer l'orientation de cette analyse. D'une part, des dispositions réglementaires et législatives nouvelles prises récemment vont modifier une partie, certes limitée, du cadre préalablement défini par la loi de 1998 et les textes d'application. D'autre part, la survenance d'événements spectaculaires ou de développements factuels qui n'avaient pas été envisagés il y a cinq ans amènent, au-delà de la stricte évaluation de la loi et des instances et mécanismes qu'elle a crée, à se demander si la sécurité sanitaire ne risque pas d'être mise en cause alors que ces textes et instances paraîtraient atteindre les objectifs qui leur ont été assignés il y a quelques années.
Enfin, une observation liminaire doit être formulée s'agissant de l'appréciation de la valeur de l'expertise qui par définition est un volet important de l'activité de l'AFSSAPS. Il ne saurait être question d'évaluer la qualité du travail scientifique car la tâche n'est pas plus accessible à l'Office qu'elle ne l'est aux deux inspections générales qui ont réalisé l'audit. Celles-ci ont précisé dans leur rapport que « cet examen a porté essentiellement sur l'organisation et les procédures internes, la mission ne pouvant se prononcer sur la qualité scientifique de la production de l'Agence ».
Le Directeur général de la santé, M. Lucien Abenhaïm, dans sa réponse (24 juin 2003) aux deux inspections générales avait fait sur ce point l'observation suivante :
« En premier lieu, le rapport indique que les auteurs ne sont pas en mesure de juger de la qualité scientifique des travaux menés par l'agence. Vous comprendrez que, dans la mesure où il s'agit du fondement même de la mission de l'agence, ce constat me rende perplexe. Le rapport porte donc plus sur des aspects de gestion que de fond, ce qui, sans diminuer son intérêt, limite sa portée et les conséquences que l'on peut en tirer pour l'avenir. La seule remarque sur la qualité globalement satisfaisante des travaux de l'agence dans ses missions de sécurité sanitaire étant celle du directeur général de la santé, je tiens à ce qu'elle apparaisse dans le rapport ».
Quelques mois plus tard, le 17 novembre 2003, en vue de l'évaluation de la loi du 1 er juillet 1998, les quatre inspections générales étaient saisies par une lettre de mission dans laquelle il était précisé notamment :
« Nous demandons aussi à la mission d'apprécier la qualité scientifique et technique de l'activité des établissements : la pertinence scientifique de leurs actions, qu'elles résultent de leur initiative propre ou qu'elles répondent à une commande de l'administration, sera évaluée tant sous un angle individuel que sous celui d'une participation globale au renforcement de la veille et de la sécurité sanitaire ».
Le rapport d'évaluation de mai 2004 comprend sur ce point, en forme de réponse, la précision suivante :
« La deuxième interrogation a porté sur le contenu de « l'évaluation de la qualité scientifique et technique des établissements » demandée dans la lettre de mission. Les experts scientifiques associés à la mission ont estimé, au terme d'une réflexion partagée avec les membres des inspections générales, qu'il ne leur était pas possible dans les délais impartis et dans le format prévu, de procéder à une évaluation de l'activité scientifique des établissements selon les méthodes et standards qui gouvernent ce type de processus ».
Le problème peut se résumer simplement dans la question « qui peut expertiser l'expertise et comment ? ». C'est l'une des difficultés qui caractérise de manière récurrente ce domaine.
On rappellera tout d'abord l'ampleur des missions de l'AFSSAPS et la structure qui a été conçue et développée pour les remplir. Les difficultés de la montée en puissance, déjà largement analysées précédemment, seront précisées et remises en perspective.
La stabilisation en vue sera appréciée en elle-même, mais aussi à la lumière des modifications institutionnelles qui peuvent la remettre en cause.
Enfin, deux aspects plus factuels déjà évoqués constitueront les deux derniers développements : d'une part les interrogations sur la position de l'Agence face à des dossiers d'actualité particulièrement sensibles, notamment au sujet de médicaments ayant fait l'objet d'un retrait, d'autre part les risques émergents.
I. UNE STRUCTURE AUX MISSIONS ÉTENDUES
L'Agence du médicament, préfiguration de l'AFSSAPS, créée par la loi n° 93-5 du 5 janvier 1993 témoignait déjà d'une volonté de conférer une autonomie certaine et des moyens importants à la hauteur des défis passés et à venir. A ces défis, identifiés, la direction de la pharmacie et l'Inspection du ministère de la santé ainsi que le laboratoire national de la santé ne pouvaient plus faire face.
On doit souligner que cette nouvelle forme d'organisation et d'exercice des compétences de l'Etat a essaimé dans d'autres champs de la sécurité sanitaire en 1998 avec l'AFSSA et l'InVS. Elle constitue donc une innovation, un précédent et un cas d'école.
1.1. Un domaine étendu
L'expérience de l'Agence du médicament s'est en effet révélée positive : dans leur rapport, MM. Claude Huriet et Charles Descours 30 ( * ) intitulaient en effet ainsi le paragraphe sur le médicament : « la sécurité sanitaire du médicament à usage humain : le bilan et les perspectives sont satisfaisants ». Cette remarque a d'autant plus de portée que les critiques faites dans d'autres domaines étaient à la fois rigoureuses et sévères (cf. supra).
Dans le cas des dispositifs médicaux précisément, l'analyse de la situation amènerait à des conclusions inquiétantes dont certaines, malheureusement, restent actuelles en raison de la faiblesse du dispositif européen qui perdure. Il s'agit d'un des domaines dont le transfert à la nouvelle agence, l'AFSSAPS, illustre l'extension des compétences par rapport à l'Agence du médicament.
L'agence sanitaire qu'est l'AFSSAPS a vu ses compétences inclure désormais, au-delà du médicament : (art. L. 5311-1 du code de la santé publique, liste non limitative) :
-- les biomatériaux et dispositifs médicaux : sous ce dernier terme, on désigne des éléments très nombreux : prothèses, orthèses, matériels et produits de diagnostic, matériel médical et hospitalier (cas de lits d'hôpital pour lesquels un grave problème de sécurité s'est notamment posé).
-- les produits contraceptifs et contragestifs,
-- les produits sanguins labiles, l'EFS (Etablissement français du sang) étant l'opérateur dans ce domaine,
-- les organes, tissus et cellules sous réserve des dispositions nouvelles contenues dans la loi du 6 août 2004 relative à la bioéthique,
-- les produits de thérapie génique et cellulaire,
-- les produits thérapeutiques annexes,
-- les réactifs de laboratoire,
-- les produits insecticides et antiparasitaires à usage humain,
-- les aliments diététiques destinés à des fins médicales spéciales,
-- les produits cosmétiques et d'hygiène corporelle.
Les travaux parlementaires et l'ensemble de la réflexion qui ont eu lieu sur la problématique de la sécurité sanitaire entre 1993 et 1998 ont convergé vers le choix d'une agence unique pour tous les produits de santé ; on verra que l'expérience ultérieure a validé à nouveau cette option qu'il convient encore de confirmer aujourd'hui. L'approche par produit dans un domaine large, mais bien défini et cohérent, parcellisant l'action de l'Etat reste donc à éviter, pour ne pas dire à proscrire. Cette remarque a d'autant plus d'intérêt que des modifications du périmètre de l'AFSSAPS ont pu être envisagées ou ont été dans certains cas réalisées.
1.2. Des pouvoirs renforcés
L'AFSSAPS étant chargée non seulement de l'évaluation du risque en matière de produits de santé, mais encore de sa gestion (ce qui la différencie fondamentalement de l'AFSSA), elle est investie de pouvoirs importants en rapport avec cette responsabilité générale. Ainsi la majorité des décisions dans son domaine de compétences exclusives sont prises par le directeur général au nom de l'Etat et non par le ministre sur avis de l'Agence. Le ministre dispose toutefois d'un pouvoir d'opposition 31 ( * ) :
« Les décisions prises par le directeur général en application du présent article ne sont susceptibles d'aucun recours hiérarchique. Toutefois, en cas de menace grave pour la santé publique, le ministre chargé de la santé peut s'opposer, par arrêté motivé, à la décision du directeur général et lui demander de procéder, dans le délai de trente jours, à un nouvel examen du dossier ayant servi de fondement à ladite décision. Cette opposition est suspensive de l'application de cette décision ».
* L'extension du champ d'intervention de l'AFSSAPS par rapport à l'Agence du médicament s'est accompagnée d'un renforcement de ses prérogatives dans les domaines nouveaux par rapport à celles que détenaient jusque là les entités administratives qui en étaient chargées.
Ainsi, un régime déclaratoire est prévu pour les établissements de fabrication, d'importation ou de distribution de matières premières destinées à l'usage pharmaceutique (avec imposition des règles de bonnes pratiques), de dispositifs médicaux et de dispositifs de diagnostic in vitro.
Le régime d'autorisation s'applique aux produits thérapeutiques annexes et aux essais cliniques portant sur les thérapies génique et cellulaire.
Il y a lieu de rappeler également que la réforme des essais cliniques qui vient d'être adoptée avec la transposition de la directive européenne sur ce sujet, confère de nouvelles compétences assez lourdes à l'AFSSAPS (cf. infra partie III). Certaines de ces nouvelles prérogatives seront d'ailleurs évoquées dans les développements relatifs à la montée en puissance et à l'objectif de stabilisation. Enfin, on n'oubliera pas que l'Agence est chargée du contrôle de la publicité de tous les produits, objets, appareils et méthodes revendiquant une finalité sanitaire.
* L'AFSSAPS dispose par la loi de 1998 de pouvoirs de police étendus qui visent tous les produits de santé qu'ils soient ou non soumis à autorisation (déclaration etc ...), et qui peuvent être mis en oeuvre dès lors qu'un produit présente ou est soupçonné de présenter, dans des conditions normales d'emploi, un danger pour la santé. Le cadre de sa fonction, donc de ses pouvoirs de police sanitaire, est très large puisqu'il va, pour tous les produits de santé, des essais cliniques à l'évaluation médico-technique et de la mise sur le marché à la vigilance sanitaire.
L'AFSSAPS a donc le pouvoir :
-- d'interdire ou de suspendre, en cas de danger pour la santé humaine, toute activité relative à un produit de santé non soumis à une procédure d'autorisation ou d'enregistrement préalable (produits cosmétiques, préparations magistrales, dispositifs médicaux, produits sanguins labiles, organes, etc),
-- d'interdire une activité illégale lorsqu'un produit est mis sur le marché sans avoir obtenu l'autorisation, la certification ou l'enregistrement préalable exigé pour ce produit,
-- de faire procéder, en cas de décision d'interdiction ou de suspension, au retrait, voire à la destruction, de tout produit dangereux.
* Les fonctions d'inspection qui préexistaient à la création de l'AFSSAPS ont été sensiblement renforcées et étendues dans le temps (suppression de toute plage horaire) et dans l'espace (tous locaux, lieux, installations et véhicules) et tout type de données. Les pouvoirs de consignation et de saisie ont été accrus, mais semblent encore insuffisants (exigence pour la saisie d'une autorisation du président du tribunal de grande instance).
1.3. Les structures de l'AFSSAPS
Pour faire face aux compétences étendues et aux responsabilités importantes qui sont les siennes, l'AFSSAPS a été dotée de moyens importants (cf. infra) sachant que son effectif atteint maintenant 940 personnes (730 en 1999) dont les deux tiers environ sont médecins, pharmaciens, scientifiques et techniciens.
* La structure interne de l'Agence a été logiquement calée à partir des catégories de produits d'une part (médicaments, dispositifs) et de fonctions d'autre part (direction des laboratoires et des contrôles, de l'inspection et des établissements, de l'administration et des finances). Toutefois, des changements substantiels ont été opérés depuis 1999 et aussi au cours des deux dernières années. Ces changements s'expliquent principalement par deux facteurs.
D'une part, la structure d'origine avait une empreinte historique qui appelait des retouches, mais plus grave, certains dysfonctionnements constatés par les inspections, en ce qui concerne l'administration générale, les finances et les systèmes d'information exigeaient une remise en ordre selon une structure plus opérationnelle. C'est ce qui a été réalisé au cours de l'année écoulée et semble répondre aux demandes des inspections et audits.
D'autre part, certains changements sont directement le produit de modifications des liens juridiques, (responsabilité du secrétariat de la transparence à la DGS en septembre 2003), y compris au niveau de la loi avec le transfert de la Commission de la transparence à la nouvelle HAS (Haute Autorité de Santé).
C'est pourquoi l'organigramme ci-après, qui est celui d'août 2004, comporte des différences sensibles avec celui du début 2003, mais il va également être modifié pour tenir compte du changement qui vient d'être évoqué (sur les conséquences éventuelles de ce changement. cf. infra partie III).
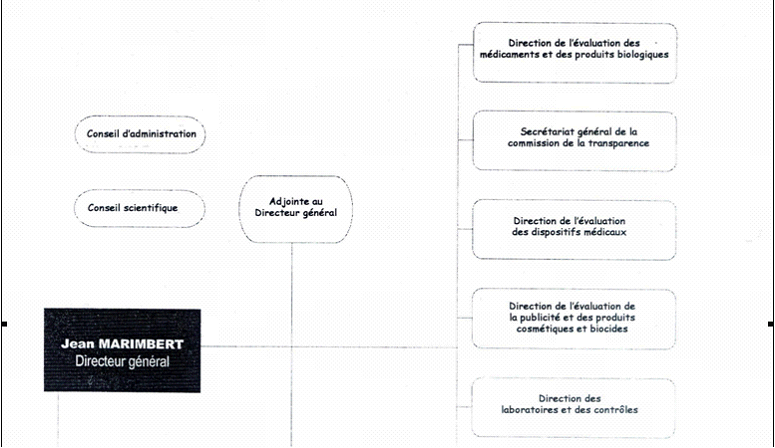

* Par rapport à la précédente structure, plusieurs remarques peuvent être faites :
-- le poste de directeur auprès du directeur général dont la justification de principe et l'efficacité dans les faits avait été contestée, a été supprimé.
-- Les trois unités de logistique administrative sont désormais sous l'autorité directe du secrétaire général. D'autres modifications importantes sont intervenues dans ce domaine : ainsi une cellule d'audit interne est en prise directe avec le secrétaire général. Seul désormais le service des affaires juridiques et européennes relève du directeur général.
-- les directions opérationnelles par produits et fonctions qui constituent en quelque sorte le coeur de métier de l'AFSSAPS ont été peu ou pas concernées par les modifications :
• évaluation des médicaments et
des produits biologiques,
• évaluation des dispositifs
médicaux,
• laboratoires et contrôles ; au
sujet de cette direction, l'audit IGF/IGAS notait (page 39) :
« Globalement, les modalités actuelles de pilotage et de
management de la DLC sont satisfaisantes »,
• inspections et établissements. L'audit
indiquait au sujet de cette dernière : « Si
l'organisation de la direction n'appelle pas d'observation, en revanche elle a
connu des difficultés en matière de recrutement. La
spécificité et les sujétions du métier d'inspecteur
ne permettent pas d'attirer de nombreux candidats en l'absence de statut
adapté ».
La direction des études médico-économiques et de l'information scientifique (DEMEIS) avait précédemment en charge le secrétariat de la commission de la transparence et donc l'interface médico-économique du médicament en particulier. La Transparence faisant depuis l'automne 2003 l'objet d'une structure qui lui est exclusivement dédiée, la DEMEIS a été transformée pour devenir la direction de l'évaluation de la publicité et des produits cosmétiques et biocides. Le secrétariat de la CEPP (commission d'évaluation des produits et prestations) a pour sa part rejoint la direction de l'évaluation des dispositifs médicaux. Les missions sur les recommandations de bonnes pratiques qui étaient partagées entre la DEMEIS et la direction de l'évaluation des médicaments et des produits biologiques (DEMEB) sont désormais toutes regroupées au sein de cette dernière.
La DEMEB avait elle-même été réorganisée dans une perspective appréciée par l'audit IGF/IGAS qui a eu lieu alors que celui-ci était en cours (op. cit. page 36-37) :
« Désormais, la DEMEB sera composée de 6 départements et de 3 services rattachés au directeur. Cette nouvelle organisation vise à donner plus de cohérence aux différentes fonctions :
-- la suppression du département des vigilances, par l'intégration des différentes unités la composant aux départements spécialisés dans l'évaluation du médicament est une mesure cohérente et pertinente.
La création d'un département de l'évaluation des produits hors AMM répond aussi à ce souci de cohérence interne en regroupant les unités en charge des ATU, des essais cliniques, des préparations hospitalières et les médicaments orphelins.
La création d'un département de l'information, de la communication et de la gestion des risques sur le médicament humain, intégrant la coordination des vigilances est encore plus nécessaire.
Actuellement, avant même la réorganisation, la coopération entre les unités de la DEMEB est de bon niveau. En effet, la synergie des travaux est assurée dans une participation croisée des différentes unités aux divers groupes de travail constitués au sein de la DEMEB. Cependant, la réorganisation envisagée devrait permettre une plus grande transversalité interne à la direction ».
Il est à souhaiter que cette structure réaménagée, qui correspond aux besoins identifiés et aux évolutions nécessaires liées aux réalités de terrain, ne soit pas rapidement remise en cause par des décisions extérieures à l'AFSSAPS.
1.4. Des moyens importants
Afin de concrétiser la volonté politique forte qui est à l'origine de la création des agences sanitaires en général, et de l'AFSSAPS en particulier, l'Etat a doté largement ces nouveaux acteurs de moyens financiers.
1.4.1. Les apports budgétaires
* Les dépenses inscrites au compte financier de l'AFSSAPS ont cru de la manière suivante :
Evaluation des charges de l'AFSSAPS (compte financier)
(en millions d'€)
|
1999 |
2000 |
2001 |
2002 |
2003 |
2004 |
|
|
Charges de fonctionnement |
44,6 |
55,8 |
56,5 |
70,6 |
86,4 |
92,16 |
|
Charges d'investissement |
50,44 |
53,2 |
60,1 |
12,4 |
4,1 |
4,56 |
Source : Direction du budget - Données du budget primitif
Il faut souligner qu'il y a eu surestimation systématique des dépenses de l'Agence dans le budget primitif, mais que cette tendance est en réduction après avoir dépassé les 20 % lors des premiers exercices significatifs. Ces surestimations ont été dues entre autres aux lenteurs du recrutement de nouveaux personnels, et à la suspension d'une opération immobilière. Les excédents d'exploitation constatés à la fin de chaque exercice ont entraîné la constitution d'un fonds de roulement considérable qui en 2002 à 37,4 millions d'euros représentait 6,4 fois le niveau jugé incompressible. La baisse des charges d'investissement témoigne de la correction opérée.
Cette situation a entraîné des remarques et des critiques tant de la part de la Cour des Comptes que des inspections générales et de la Commission des finances du Sénat (rapport d'information de M. Adrien Gouteyron n° 404 (2002-2003) du 16 juillet 2003. L'AFSSAPS : sortir de la crise de croissance (pages 49 et 50) :
« Il paraît de meilleure gestion de ramener le fonds de roulement de l'Agence à un niveau plus raisonnable, les établissements publics n'ayant pas à se constituer de telles réserves de trésorerie. (...)
Votre rapporteur croit néanmoins que l'aisance financière de l'AFSSAPS a pu, en partie, expliquer l'absence de comptabilité analytique en son sein, lui faisant négliger les réformes à entreprendre pour atteindre une réelle performance dans sa gestion ».
Enfin, le poids important de la gestion dans le domaine immobilier est également noté en des termes dépourvus d'ambiguïté :
« Or, les ambitions immobilières de l'Agence ont été contrariées, en grande partie en raison des carences de ses propres services administratifs et juridiques, et ont dû être, sinon abandonnées, du moins repoussées à une échéance inconnue. L'IGF et l'IGAS ont d'ailleurs indiqué à votre rapporteur que « toute la politique immobilière de l'Agence est à revoir ! »
Ces difficultés s'inscrivent dans le cadre de l'insuffisance de la gestion administrative et financière qui est abordée dans la partie II de la partie du présent rapport consacré à l'AFSSAPS (cf. infra).
1.4.2. L'apport des industriels
* La part des taxes, droits et redevances dans les recettes de l'AFSSAPS est un élément spécifique à cette Agence qui doit entrer en ligne de compte dans l'appréciation de sa situation financière passée, actuelle et surtout à venir (les dispositions relatives aux domaines nouveaux pour elle sont entrés en vigueur progressivement (2002). La part de l'ensemble de ces taxes en pourcentage des recettes de fonctionnement s'est élevée de 50,8 % en 2000 à 51 % en 2002 et à 82 % en 2003. Corrélativement, l'ensemble des subventions d'exploitation du budget de l'Etat à l'AFSSAPS a évolué ainsi en pourcentage des recettes de fonctionnement :
|
1999 |
2000 |
2001 |
2002 |
2003 |
|
35,1 % |
40,5 % |
24,5 % |
22,1 % |
8 % |
Cette évolution est quelque peu chaotique, mais s'explique clairement par les facteurs qui viennent d'être évoqués auxquels s'ajoute en 2003 « la volonté des tutelles d'assécher le fonds de roulement de l'établissement » (rapport précité de M. Adrien Gouteyron au nom de la Commission des finances du Sénat (juillet 2003). Evoquant le niveau de ces subventions de l'Etat à l'AFSSAPS, le rapport des quatre inspections précité (mai 2004) note (volume I, page 16) :
« Pour l'AFSSAPS, cette part varie considérablement, de 8 à 40 % sur la période, au rythme des régulations budgétaires et des augmentations des tarifs de redevance. Le niveau de 8 % est sans doute, en tendance, excessivement bas, sauf à installer l'idée que l'AFSSAPS a plus de comptes à rendre à l'industrie qui la finance qu'à l'Etat ».
Il apparaît clairement qu'il serait malsain que le financement de l'AFSSAPS relève complètement des industries concernées au-delà de ce que la logique administrative et même politique voudrait. Un certain nombre de tâches d'expertise générale de veille scientifique et technologique, à commencer par la pharmacovigilance et la totalité des inspections relèvent de la responsabilité de l'Etat et à ce titre celui-ci doit y participer à due concurrence.
Cette très rapide évocation des moyens consacrés à l'AFSSAPS illustre déjà la nécessité de ne pas changer, encore moins bouleverser, le périmètre de compétences, ce que les quatre inspections ont désigné par l'expression « un paysage institutionnel en continuelle évolution ». C'est là une analyse qui s'est rapidement imposée dès le début de l'évaluation à laquelle il est procédé ici pour tous les domaines couverts par la loi de 1998. On verra plus loin que cette mise en garde est pour le moins fondée.
II. UNE MONTÉE EN PUISSANCE DIFFICILE
Si L'AFSSAPS a succédé à l'Agence du médicament, son périmètre d'activité est néanmoins beaucoup plus large ; les conditions d'exercice de ses compétences dans le domaine du médicament lui-même ont évolué et évoluent encore. Il ne s'agit donc pas seulement d'assurer une continuité, mais de remplir des objectifs, plus lourds, plus ambitieux et plus changeants que ceux qui étaient naguère fixés à l'Agence du médicament. En outre, il y a lieu de rappeler qu'en mars 1999, l'Agence du médicament elle-même n'avait pas encore réalisé son plein développement en termes d'effectifs et de missions. Les moyens importants qui ont été donnés à l'AFSSAPS ne peuvent à eux seuls permettre l'atteinte immédiate de tous les objectifs qui lui ont été fixés, alors même que les environnements administratif, scientifique, européen se modifient à un rythme accéléré.
2.1. Les constats antérieurs
Le constat fait par la Cour des Comptes sur les premières années de fonctionnement (jusqu'en 2001) comprenait des critiques nombreuses et notamment sur la gestion interne de l'Agence, reconnaissant aussi la difficulté du contexte juridique et administratif qui s'imposait à elle ; ce constat notait aussi « la réussite attestée, par la communauté internationale, du haut niveau et de la rigueur de l'expertise scientifique de l'AFSSAPS ».
L'audit IGF/IGAS, achevé en décembre 2002, en examinant chacune des activités et modalités de fonctionnement de l'Agence, a porté des appréciations sévères et ciblées qui visent principalement la gestion et le fonctionnement interne de l'Agence ; la conception de l'expertise pratiquée par l'Agence y fait l'objet d'interrogations. Les résultats des activités de l'Agence sont appréciés diversement selon les domaines : lacunes (pharmacovigilance entre autres) et réussites (démarche d'assurance qualité établie par la direction des laboratoires et des contrôles).
De son côté, M. Adrien Gouteyron concluait l'avant-propos de son rapport d'information en juillet 203 dans les termes suivants : « Votre rapporteur a pu observer un établissement à qui des tâches très importantes et à haute responsabilité ont été confiées, confronté à une montée en charge rapide ayant par conséquent engendré des dysfonctionnements, certes nombreux, mais qui sont avant tout, semble-t-il, le reflet de « problèmes de croissance ».
De fait, l'Agence a accompli un travail considérable en quelques années seulement, et se réforme progressivement, tout en menant à bien les missions qui lui ont été confiées par le législateur. Il convient donc de lui laisser du temps pour s'approprier ses missions nouvelles et pour adopter une culture professionnelle rénovée ».
L'ensemble de ces analyses comprend d'abord un rappel des obstacles extérieurs à l'action de l'Agence que constituent un environnement juridique et administratif stabilisé tardivement et le problème de l'exercice de la tutelle.
2.1.1. Un environnement juridique et administratif tardivement stabilisé
Les dispositions réglementaires prévues par la loi ont été prises souvent très tardivement ; ainsi au début de l'année 2003, la publication de cinquante textes d'application était encore attendue dans les domaines les plus divers, mais particulièrement dans ceux relatifs aux nouvelles compétences de l'Agence : cosmétiques, dispositifs médicaux, organes cellules et tissus issus du corps humain, produits thérapeutiques annexes (produits entrant en contact avec les organes, tissus et cellules), matières premières à usage pharmaceutique. A l'automne 2004, la situation était normalisée pour l'essentiel. Outre les textes prévus par la loi, d'autres dispositions peuvent, par leur absence, poser de réels problèmes à l'Agence : cas de l'obligation déclarative pour les préparations magistrales et officinales par exemple ou celui des mesures destinées à prévenir les ruptures de stock de médicaments dans la chaîne de distribution ; en ce qui concerne ce dernier point, l'AFSSAPS a instauré d'elle-même un dispositif très efficace de signalement qui a précédé de plusieurs années la disposition incluse tout récemment à cette fin dans la loi de santé publique du 9 août 2004.
Les raisons de ces retards résident principalement dans la longueur et la complexité des procédures de rédaction des textes avec plusieurs intervenants dont la DGS, elle-même très chargée, et les instances européennes où les délais excessivement longs peuvent s'expliquer par de graves divergences de fond sur le degré de sécurité sanitaire (cf. infra).
A côté des retards enfin comblés ou en voie de l'être, des lacunes peuvent perdurer (cf. supra). En témoigne la tentative de créer l'obligation de communication des formules des produits cosmétiques à l'AFSSAPS lors de l'examen du projet de santé publique, obligation dont l'absence constitue un obstacle à la vigilance vis-à-vis de ces produits.
D'une manière générale, pour faire face à ces insuffisances de textes et aux difficultés que l'Agence rencontre dans des « compétences frontières » où d'autres instances exerçant leur activité (AFSSA, InVS, ANAES), l'AFSSAPS a précisément crée en son sein un comité de coordination des vigilances, auquel s'ajoutent d'autres mécanismes spécifiques qui témoignent d'un pragmatisme généralement efficace.
Enfin, au-delà des retards et des lacunes passés ou actuels, on peut craindre que la relative stabilisation de l'environnement réglementaire soit remis en cause par de nouvelles dispositions législatives récentes qui ne s'inscrivent pas dans le cadre fixé en 1998 (cf. infra).
2.1.2. L'exercice délicat de la tutelle sur l'AFSSAPS
Le ministère chargé de la santé exerce la tutelle de l'Agence, même si on relève aussi au sein du conseil d'administration un représentant de la Direction du budget, de la DGCCRF (Direction générale de la concurrence du contrôle et de la répression des fraudes), ainsi que de la Direction générale de l'industrie.
Deux directions, au sein du ministère de la santé, participent à l'exercice de la tutelle : la DAGPB (Direction de l'administration générale, des personnels et du budget) et à titre principal la DGS, elle-même à travers deux sous directions : la 3 ème pour les aspects techniques et les orientations relèvant de la politique de la santé et la 4 ème pour les aspects méthodologiques de mise en place de nouveaux moyens d'exercice de la tutelle (préparation du contrat d'objectifs et de moyens).
L'exercice de la tutelle est donc naturellement rendu plus complexe. On aurait pourtant pu espérer qu'avec un seul ministère de tutelle (contrairement à l'AFSSA où il y en a trois), les choses auraient été plus simples et les relations moins problématiques. Cela étant, des efforts ont été faits des deux côtés pour rationaliser les relations et on peut ainsi considérer qu'une partie des difficultés a été résolue. Au titre d'un accord passé en 2002 entre la DGS et a DAGPB pour l'ensemble des agences sanitaires, il a été confirmé que la DGS est la direction « chef de file » en charge de la définition des orientations stratégiques. D'autre part, l'AFSSAPS a mis en application un système de suivi de l'ensemble de ses échanges avec la tutelle.
Il reste que la situation est naturellement délicate pour une direction générale qui exerce la tutelle d'orientation, notamment scientifique, mais qui n'a plus à sa disposition les experts pour pouvoir contrôler les activités scientifiques de l'Agence. Il convient de rappeler que le directeur général de l'AFSSAPS détient dans tous les domaines de compétence de l'Agence le pouvoir de police sanitaire (décision de retrait d'un médicament par exemple).
Ce n'est que sur la question de l'admission du remboursement par la sécurité sociale que le ministre, à partir de l'avis de la commission de la transparence, dispose du pouvoir de décision. Cet ordonnancement va d'ailleurs disparaître avec la création de la HAS (Haute Autorité de Santé) par la loi du 13 août 2004 relative à l'assurance maladie (cf. infra). En outre, la DGS s'est souvent sentie tenue à l'écart (« court-circuitée ») par le fait que dès la création de l'AFSSAPS, ses directeurs généraux ont entretenu des relations privilégiées et directes avec le cabinet du ministre chargé de la santé, ce que soulignent tous les rapports précités (Cour des Comptes, inspections générales, rapporteur de la commission des finances du Sénat).
L'affaiblissement du pouvoir de la DGS a réduit l'exercice de la tutelle, le pilotage stratégique de l'Agence et d'abord la gestion interne. De ce fait certaines insuffisances ont pu perdurer.
2.2. Les insuffisances et les progrès soulignés
Les questions relatives à la structure d'ensemble et aux moyens généraux de l'Agence ont été abordées dans le développement précédent du rapport.
Les travaux d'audit, d'inspection ou d'analyse ont dégagé un ensemble de critiques, dont certaines assez graves, qui pour l'essentiel convergent. Ces constatations de terrain doivent être brièvement rappelées sachant qu'elles s'appuient sur un examen détaillé.
2.2.1. Les insuffisances des fonctions support
Deux secteurs ont présenté depuis le début de graves insuffisances : les systèmes d'information et la direction de l'administration et des finances. L'audit (IGF-IGAS) a même considéré que « les dysfonctionnements des fonctions support sont de nature à compromettre le bon accomplissement des missions de sécurité sanitaire » (page 47).
2.2.1.1. Les systèmes d'information
L'importance exceptionnelle des applications informatiques dans les domaines d'activité de l'AFSSAPS n'est pas à démontrer. Ce que représente un dossier d'AMM à lui tout seul en volume et complexité est suffisamment parlant. L'extrême diversité des domaines abordés en raison des compétences très larges de l'AFSSAPS et la confidentialité de nombre de données accroît encore les difficultés. L'Agence a ainsi recueilli des applications anciennes, peu performantes, obsolètes sans parler de l'absence de réseau entre des applications qui sont destinées à être partagées.
L'AFSSAPS ayant pris progressivement la mesure des efforts à accomplir pour ne pas dire des chantiers à ouvrir, des impulsions ont été données depuis deux ans pour redresser la situation. Ainsi, des décisions prises en octobre 2002 ont ouvert des perspectives concrètes dont le redéveloppement de l'application de pharmacovigilance ainsi que de l'application de gestion des autorisations temporaires d'utilisation et la poursuite des efforts en matière de production automatisée de document normalisés (résumés des caractéristiques produits, avis de la transparence).
En outre, des moyens importants, humains notamment, ont été consacrés depuis trois ans au département des systèmes d'information et de documentation. La standardisation des matériels et la constitution des réseaux indispensables sont réalisés ou en cours.
Les projets annoncés en avril 2003 se mettent progressivement en place : pharmacovigilance, suivi de dossiers, communication électronique avec l'extérieur.
Par ailleurs, le projet @MM doit enfin constituer la banque de données médicaments attendue puis longtemps. Une explication avec un retour sur un épisode précédent est ici nécessaire.
La nécessité de disposer d'une base d'information, de référentiels de bonne pratique, de prescription et d'usage du médicament par les autorités publiques responsables de la sécurité sanitaire est une évidence. La mise en place de tels outils et surtout leur ancrage administratif ne se sont pas imposés, à l'origine du moins, avec la même évidence. La multiplicité des intervenants est une source de difficultés qui risque de s'aggraver encore (cf. infra).
La loi de financement de la sécurité sociale pour 2001 a notamment fixé comme objectif à l'AFSSAPS la réalisation d'une base de données administratives et scientifiques sur les médicaments et les dispositifs médicaux devant constituer la référence pour les professionnels de santé et les administrations. L'AFSSAPS a ainsi conçu le projet @MM qui consiste en la diffusion sur le site Internet de l'Agence des décisions d'autorisation de mise sur le marché et des indications complémentaires, notamment la notice réglementaire destinée aux patients.
Pour ce projet qui devait être réalisé au 1 er janvier 2003, l'Agence a été amenée à mobiliser d'importants moyens et les restructurations internes (direction de l'évaluation des médicaments et des produits biologiques) ont été orientées dans cette perspective. L'échéance n'a pas été tenue exactement puisqu'une première version réduite a d'abord été mise en place en juin 2002. Plusieurs séries d'obstacles ont été à l'origine de cette situation : fiabilité discutable de certaines des données préexistantes, hétérogénéité des documents de base et de leurs formats, faiblesse déjà signalée de l'ensemble des moyens informatiques de l'AFSSAPS.
Les efforts faits par l'Agence ont permis d'approcher progressivement l'objectif fixé en 2001 comprenant systématiquement le RCP (résumé des caractéristiques du produit), dans une base de données structurée et homogène, le flux des nouvelles demandes étant maîtrisé, mais le rattrapage des anciennes AMM restant à régler.
2.2.1.2. La direction de l'administration et des finances
L'importance des moyens budgétaires dont l'AFSSAPS a bénéficié depuis sa création ne s'est pas accompagnée d'une structuration administrative en rapport : c'est plutôt le contraire qui a été constaté par la mission d'audit des deux inspections générales à la fin de l'année 2002 dans des termes d'une très grande sévérité. La situation de la direction de l'administration et des finances a été ainsi qualifiée de « sinistrée ».
Parmi les manquements les plus graves pointés au cours de la période 2000-2002, sont signalés :
- pour les contrôles en laboratoires : interruptions des commandes de matériels et produits pendant six mois (octobre 2000-mars 2001) avec conséquences sur le fonctionnement des laboratoires, leur crédibilité au niveau européen en particulier, de même pour le non-renouvellement de contrats de maintenance de matériels scientifiques ;
- pour la rémunération des experts extérieurs, des délais de paiement atteignant de 15 à 27 mois de retard ;
- les conséquences d'une gestion très insuffisante sur la politique et les actions en matière immobilière ;
et sur un plan plus général :
- l'absence de procédure budgétaire rationnelle, de comptabilité analytique et de moyens de contrôle de gestion.
Il n'est donc pas étonnant que les préconisations de la mission d'audit sur la direction de l'administration et des finances se soient conclues dans les termes suivants 32 ( * ) :
« Enfin, au vu des considérations précédentes et du diagnostic porté par la mission, il est hautement souhaitable dans une logique de dynamique nouvelle à faire partager à l'ensemble des équipes, d'appeler à la tête de cette direction, dans les meilleurs délais, un nouveau directeur ».
Il semble que ce diagnostic dépourvu de ménagement ait eu des suites concrètes. En outre, ont été réalisés depuis le début de l'année 2004 la mise en place d'une comptabilité analytique, une fonction d'audit interne et un contrôle de gestion avec ainsi une formalisation des procédures entre les directions techniques et la direction de l'administration et des finances. La fonction « achats » a été réorganisée avec, comme il convient, une comptabilité générale d'engagement. Une logistique d'achat des laboratoires a été reprise et intégrée à la direction des laboratoires, ce qui a permis de mettre fin aux blocages constatés précédemment.
Symbole de cette remise à niveau de la gestion interne, le COM (contrat d'objectifs et de moyens) devrait enfin pouvoir être passé dans les premiers mois de l'année 2005.
2.2.2. Les insuffisances de l'orientation stratégique
* Outre l'absence particulièrement symbolique du COM qui vient d'être rappelée, l'organisation de la direction générale a présenté jusqu'en 2003 une structure peu rationnelle et mal adaptée qui finissait par constituer un obstacle à la détermination des orientations stratégiques à une agence de l'importance de l'AFSSAPS. La mission d'audit relevait ainsi 33 ( * ) :
« Dans cette organisation, le Directeur général est assisté d'un secrétaire général, d'un directeur auprès du Directeur général, dont les missions réelles respectives sont mal définies :
- le secrétaire général n'a ni une compétence d'ensemble sur les moyens de l'organisme, ni une compétence transversale de « numéro 2 » de l'Agence. La supervision du service des affaires juridiques tout comme celle des affaires financières (directement rattachées au DG) lui échappe.
- la direction auprès du Directeur général regroupe en son sein des fonctions éparses (communication, Internet, affaires communautaires, affaires juridiques, secrétariat des conseils ...), dans un périmètre transversal fréquemment modifié ».
L'ordonnancement de cette structure a été sensiblement amélioré avec la suppression du poste de direction auprès du directeur général et l'extension de la compétence d'ensemble du secrétaire général à toutes les fonctions support (administration et finances, ressources humaines, systèmes d'information).
* Une organisation bien adaptée et la perspective de l'établissement d'un COM sont des conditions nécessaires au pilotage stratégique attendu, mas d'autres éléments doivent être recueillis pour que les conditions suffisantes soient réunies. On ne détaillera pas les éléments d'appréciation de la gestion et des indicateurs qui ont été développés depuis près de deux ans, les améliorations les plus significatives ayant déjà été mentionnées. On peut indiquer toutefois que l'absence d'éléments d'appréciation de l'adéquation des moyens aux missions a des conséquences regrettables qui freinent encore l'optimisation des moyens de l'Agence. En revanche, il convient de rappeler et de citer quelques difficultés d'ordre général qui restent à résoudre.
- Des progrès restent à accomplir dans les relations avec la tutelle de la DGS. Ses insuffisances pèsent sur le choix de certaines orientations de l'Agence d'autant que le transfert de la commission de la transparence à la Haute Autorité de Santé risque de constituer un élément de perturbation ;
- Les rôles du conseil d'administration et du conseil scientifique méritent d'être sérieusement précisés, notamment par leur participation aux orientations stratégiques. Ils n'ont pas été saisis de ces questions, tout du moins jusqu'à une période récente. Le rôle du conseil scientifique semble avoir été limité essentiellement à l'examen des subventions de recherche allouées par l'Agence.
2.2.3. Le nouvel équilibre des activités de l'Agence
L'équilibre entre les différentes missions de l'Agence fait l'objet de critiques multiples, mais qui peuvent néanmoins appeler des appréciations nuancées. C'est la détermination des priorités qui est ici en cause. Or, au-delà de grilles d'appréciation bien construites, on peut également considérer que le rôle dévolu « aux urgences » ne doit pas être excessivement réduit ab initio, faute de quoi l'actualité pourrait prendre sa revanche en cas de crise. C'est pourquoi les critiques souvent faites sur un nécessaire rééquilibrage entre les missions et activités peuvent elles-mêmes faire l'objet d'un examen.
Il a été notamment reproché à l'AFFSAPS de consacrer trop de moyens aux missions traditionnelles héritées de l'Agence du médicament, à savoir les AMM, au détriment d'autres missions (secrétariat de la commission de la transparence, cosmétiques, dispositifs médicaux). Il n'est pas aisé et est même souvent impossible d'apprécier avec précision la justesse de ces critiques qui reposent sur la connaissance de données nombreuses et complexes à interpréter ; en revanche, certaines d'entre elles peuvent être efficacement remises en perspective pour les approuver ou au contraire les contester.
2.2.3.1. La part des procédures d'AMM
L'instruction des dossiers et les procédures d'examen des demandes d'autorisation de mise sur le marché des médicaments constituent « le coeur de métier » de l'AFSSAPS quelle que soit la procédure suivie : nationale, dans le cadre européen, de reconnaissance mutuelle ou centralisée ; dans ce dernier cas, c'est l'EMEA (Agence européenne d'évaluation des médicaments) qui charge l'AFSSAPS d'un dossier.
Il apparaît clairement que l'AFSSAPS remplit ses missions. La mission d'audit elle-même a porté une appréciation qui ne laisse pas de place au doute dans l'appréciation de la valeur et de l'efficacité de l'activité de l'Agence (op. cit. page 9) :
« Une procédure satisfaisante mais perfectible
Ce schéma général de l'instruction des demandes d'AMM apparaît satisfaisant à la mission. La mission ne peut pas se prononcer sur le fond des dossiers d'évaluation, ne disposant pas de compétence ni de légitimité médicale. Néanmoins, les procédures et l'organisation en matière d'évaluation du médicament sont maîtrisées.
En effet, la multiplicité d'intervenants dans cette procédure semble être une force du système d'évaluation. L'avis de très nombreux spécialistes permet d'aboutir à une conclusion la plus objective possible.
Par ailleurs, la nature des décisions prises à l'issue de l'instruction des nouvelles demandes nationales ou de reconnaissance mutuelle est un élément d'appréciation de l'activité. Ainsi en 2001, 42 % de ces décisions ont fait l'objet d'une suspension de la procédure dans l'attente d'informations complémentaires demandées aux laboratoires, et 15,7 % d'entre elles d'un refus d'autorisation.
Enfin, l'implication de l'Agence dans les procédures européennes est satisfaisante. L'AFSSAPS instruit toutes les nouvelles demandes en procédure centralisée. Le nombre d'évaluations produites en tant qu'Etat destinataire est en augmentation (68 en 2000 et 89 en 2001). L'instruction des variations de type II en procédure centralisée ou en reconnaissance mutuelle est également systématique, avec un degré d'investissement décidé au cas par cas.
Une nuance doit être apportée à ce tableau tout à fait positif en ce qui concerne les délais d'instruction pour les procédures nationales. En effet, l'Agence doit respecter les limites contraignantes fixées pour les procédures européennes (210 jours en procédure centralisée, 90 jours en reconnaissance mutuelle). Les procédures nationales sont traitées en seconde priorité et ont connu des retards importants, passant de 124 jours en 2000 à 205 jours pour les neuf premiers mois de 2002. Il en est de même pour les dossiers d'extension d'indications et de vaccinations thérapeutiques. Toutefois, comparer pour ces dernières, un délai de 150 jours (qui doit indiscutablement être sensiblement réduit) à un délai de 30 jours prévu par les dispositions européennes montre que ce dernier chiffre n'est pas réaliste. On peut s'inquiéter quant à la philosophie qui sous-tend un tel dispositif.
Dans sa lettre du 24 juin 2003 contenant ses observations sur le rapport d'audit, M. L. Abenhaïm contestait l'équilibre des moyens consacré aux différentes missions de l'AFSSAPS dans les termes suivants :
« Sans remettre en cause la qualité globale des décisions prises par l'agence, j'avais indiqué à la mission que je m'interroge beaucoup sur la pertinence de la répartition des moyens de l'agence entre ses missions :
- Importance de l'investissement sur la procédure d'octroi des AMM, en particulier sur la procédure centralisée.
- Apport insuffisant de moyens pour les commissions placées auprès du Ministre et dont elle assure le secrétariat, comme la commission de la Transparence ».
« Sur ce point aussi, le rapport ne se prononce pas et ne permet pas de disposer d'éléments factuels faisant le point avant la négociation du contrat d'objectifs et de moyens. (...)
S'agissant de l'implication de l'agence dans les procédures européennes d'AMM, je regrette que le rapport ne se prononce pas sur son niveau au regard des performances dans d'autres secteurs de l'activité de l'agence. On peut se demander si son degré est justifié. En d'autres termes, le rapport devrait poser la question de savoir si le maintien d'un haut niveau de compétence au sein de l'agence justifie qu'elle consacre à ces tâches une telle quantité de ressources humaines alors que dans le même temps, les ressources en matière d'évaluation font si cruellement défaut au secrétariat scientifique de la commission de la transparence. De plus, il faut souligner que ni l'AFSSAPS, ni l'EMEA, n'effectuent d'audit des données des études comme le fait la FDA, du fait de la qualité de ses ressources internes. En d'autres termes, le travail en reste au niveau de « l'opinion » d'expert, ce qui est largement insuffisant, et pour l'AMM et pour la Transparence. En général, ce processus qui est presque uniquement basé sur les données des laboratoires, est par nature insatisfaisant ».
On peut noter au passage que la référence aux travaux de la FDA, critique à l'égard de l'AFSSAPS et de l'Agence européenne, ne paraît pas corroborée par les révélations récentes sur la valeur des travaux de l'Agence américaine pour des autorisations de mises sur le marché préalablement déposées aux Etats-Unis, ni pour le suivi de ces médicaments après une première autorisation (cf. infra en particulier le cas du Vioxx et celui de la cérivastatine).
De son côté, M. Adrien Gouteyron, sénateur, dans son rapport d'information à la commission des finances (page 57), notait en juillet 2003 :
« Une trop grande priorité accordée aux missions traditionnelles de l'Agence
La Cour des comptes, au moment de son contrôle, c'est-à-dire entre la fin 2000 et le début de l'année 2001, avait jugé que l'AFSSAPS n'avait pas encore pris totalement le contrôle de son champ de compétences, ce qui avait notamment pour conséquence de handicaper la fixation des prix des médicaments par la commission de la transparence.
L'Agence avait surtout donné la priorité aux aspects qui engagent sa responsabilité alors que, dans les autres domaines, les produits cosmétiques par exemple, les moyens mis en oeuvre l'avaient été moins rapidement. De même, l'évaluation des dispositifs médicaux n'était pas en place alors que ce poste de dépenses a augmenté de 16 % en 2002.
Lors de son audition, le directeur général de la santé a fait le même constat. Il a, en effet, estimé que les moyens alloués par l'AFSSAPS aux autorisations de mise sur le marché sont beaucoup plus importants que ceux affectés à la commission de la transparence, alors que les AMM sont aujourd'hui très largement dictées par lé réglementation communautaire et délivrées par l'Agence européenne du médicament ».
Pour en rester à la seule place des procédures d'AMM dans l'ensemble des activités de l'Agence, il apparaît difficile de se satisfaire de la bonne implication de l'Agence dans les procédures européennes, de la consistance de ses travaux et méthodes, et de lui reprocher par ailleurs de consacrer trop d'énergie à ces missions essentielles, notamment « celles qui engagent sa responsabilité ». Sa responsabilité est d'ailleurs engagée aussi pour des domaines moins spectaculaires que sont les dispositifs médicaux et les cosmétiques.
Le souci de « tenir son rang » et de constituer une référence dans le concert des agences nationales en étroite liaison avec l'Agence européenne (EMEA), ce qui a précisément été assuré et relevé par les instances d'inspection, paraît au contraire tout à fait positif. L'apparition de risques nouveaux, dont les développements se précisent rapidement au niveau européen et mondial, justifient pleinement la recherche d'une vigilance et d'une efficacité au meilleur niveau. A titre d'illustration, pour le contrôle des médicaments sous AMM européenne centralisée, la France est, après l'Allemagne, le deuxième pays de l'Union européenne le plus souvent choisi pour effectuer ces contrôles.
Non seulement la place prise par les dispositions communautaires ne doit pas amener à affaiblir les dispositifs qui assurent un haut niveau de sécurité sanitaire, mais encore les risques d'une concurrence vers le bas entre agences, et l'existence de mécanismes communautaires contestables sur la sécurité des dispositifs médicaux ou de conditions inquiétantes de dispensation des médicaments par exemple, sans parler de la valeur des contrôles des laboratoires, apportent une justification supplémentaire aux efforts consacrés par l'Agence à ces activités. Les recommandations de la mission d'audit (rapport p. 60) se plaçant dans une perspective plus générale encore vont précisément dans le sens d'une implication européenne encore plus grande de l'AFSSAPS en renvoyant sans ambiguïté la responsabilité des décisions à la tutelle, c'est-à-dire au ministère chargé de la santé et donc à la D.G.S. :
« Engager une réflexion sur l'implication de l'Agence dans les travaux au sein de l'Union européenne.
Au regard de l'importance croissante des moyens consacrés aux activités européennes, même si la mission n'a pu les quantifier précisément dans toutes les directions, il est aujourd'hui important que cette question soit placée au coeur des débats entre l'Agence et ses tutelles.
Au-delà de la participation de l'Agence à des procédures européennes telles que les procédures d'AMM centralisées, le discours général de l'Agence justifiant son implication à l'échelle européenne, porte sur le niveau de sécurité sanitaire souhaité à terme au sein de l'Union.
De plus, comme la mission a pu le montrer, de réels sujets d'inquiétude peuvent d'ores et déjà être identifiés dans l'Union, compte tenu de la liberté de circulation des produits et de l'absence d'harmonisation effective des pratiques de sécurisation des produits de santé entre Etats. Des propositions de la France sur ces questions seraient de nature à accroître l'investissement européen de l'Agence.
La réponse explicite des pouvoirs publics sur cette question est naturellement déterminante sur les modalités de fonctionnement de l'Agence et sur ses priorités de travail à court et moyen terme. Au vu d'une identification précise des moyens actuellement dévolus aux travaux de nature européenne par l'Agence et d'une prévision d'activité graduée selon plusieurs scenarii, les autorités de tutelle doivent être en mesure de statuer sur le degré d'investissement européen de l'Agence ».
Ajoutons que le rapport d'évaluation de la loi du 1 er janvier 1998 des quatre inspections ne reprend en rien des critiques relatives au poids excessif des activités traditionnelles de l'AFSSAPS.
Pour terminer par un exemple spécifique en matière d'autorisation, mais important par ses implications de sécurité, le cas des ATU (autorisations temporaires d'utilisation) mérite d'être cité. L'ATU est une procédure autorisant dans un but thérapeutique (et non de recherche) l'utilisation de médicaments ne bénéficiant pas d'AMM. Le rapport d'audit (page 12) précise sur ce point :
« La gestion et le suivi des autorisations d'importation appellent un jugement particulièrement favorable de la part de la mission. L'effort de formalisation des processus d'autorisation mérite d'être souligné et s'est notamment traduit par des délais de traitement (entre 24 et 48 heures) systématiquement respectés pour les importations, même si pour les autorisations d'exportation, dont le nombre progresse, ils demeurent trop longs ».
2.2.3.2. Les missions de l'AFSSAPS dans les autres domaines
- L'évaluation des dispositifs médicaux
L'Agence exerce ses (nouvelles) responsabilités dans ce domaine dans un cadre réglementaire unifié fixé par trois directives européennes qui procèdent d'une « nouvelle approche » de la sécurité basée sur la conformité des exigences essentielles attestée par un label (marquage CE). Avec la libre circulation des produits en Europe, la sécurité repose d'abord sur la responsabilité du fabricant puisque l'autorité sanitaire ne se prononce pas préalablement à la mise sur le marché. Dans ce cadre, l'élaboration des normes et de référentiels est indispensable. Les exigences essentielles que doivent respecter les fabricants restent très générales et seule l'existence de normes techniques peut permettre de les renforcer en les adaptant aux spécificités des produits. Les normes précisent ainsi les obligations sur le choix des matériaux, les performances attendues des dispositifs et les essais de vérification. Cette activité est confiée au Comité européen de normalisation et en France à l'AFNOR. Pour l'AFSSAPS, un plan de pilotage de cette activité en partenariat avec l'AFNOR est d'autant plus nécessaire que la « nouvelle approche » peut receler des risques ; l'AFSSAPS a prouvé récemment que sa vigilance dans le contrôle des dispositifs médicaux répondait aux exigences de sécurité (lentilles cornéennes et liquides d'entretien, lits d'hôpitaux).
- Les produits immunologiques (vaccins) et les médicaments dérivés du sang.
Cette activité, cadrée dans un délai de 60 jours par les dispositions européennes, a fortement augmenté entre 1999 et 2001.
- Les produits cosmétiques
Les difficultés rencontrées dans ce nouveau domaine sont essentiellement dues à une insuffisance du dispositif juridique qui prévoit que les formules de composition des cosmétiques « sont à la disposition de l'Agence » et non obligatoirement communiquées à celles-ci. La tentative récente de régler ce problème par voie d'amendement n'ayant pas abouti, il convient néanmoins de surmonter cet obstacle, faute de quoi l'AFSSAPS n'a pas les informations nécessaires pour se prononcer sur son innocuité. Il y a lieu de noter que les « centres anti-poisons » disposent quant à eux des formules de composition.
- La surveillance du marché
Le contrôle des médicaments chimiques a été affecté par la montée en puissance des contrôles sur les produits immunologiques selon la mission d'audit (rapport p. 16-17) qui détaille par ailleurs la place prise par les médicaments génériques dans le déploiement de l'activité de l'AFSSAPS :
« Par ailleurs, au sein même des médicaments chimiques, l'Agence a affiché dès 1999 une priorité pour le contrôle des médicaments génériques. Or, si ces contrôles ont représenté 78,9 % des contrôles sur les médicaments chimiques en 1999, ils n'en représentaient plus que 42,9 % en 2000 et 35,25 % en 2001. Ainsi, le nombre de spécialités génériques contrôlées est passé de 477 en 1999 à 106 en 2001. L'objectif affiché ne semble plus se traduire quantitativement dans la réalité, même si l'Agence a souhaité récemment mettre en place un programme de contrôles plus ciblé que systématique.
Combinée à la baisse globale du nombre des contrôles sur les médicaments chimiques, cette tendance pourrait être préoccupante. Aussi, convient-il qu'elle soit bien maîtrisée par l'Agence, à travers un ciblage de ses programmes de contrôles des médicaments chimiques (génériques ou pas), en fonction des informations scientifiques dont elle peut disposer.
Cependant, la mission a pu relever que les taux de non-conformités sont supérieurs pour les génériques à ceux relevés sur les princeps (en 2001, respectivement 11,4 % contre 3,7 %). Cette tendance, qui peut en partie s'expliquer par le fait que l'Agence effectue des contrôles « ciblés » semble néanmoins justifier qu'un effort particulier soit maintenu sur le contrôle des génériques ».
- L'inspection des établissements, tâche traditionnelle de l'Agence, dispose d'une méthodologie éprouvée et efficace, mais a dû faire face à de nouvelles obligations, notamment pour les produits sanguins labiles et les dispositifs médicaux.
Le nombre d'inspections réalisées a ainsi fortement augmenté, mais un retard s'est accumulé en ce qui concerne les réinspections périodiques des laboratoires pharmaceutiques.
Le fonctionnement satisfaisant du département des alertes est par ailleurs à noter : bonne réactivité pour le rappel des lots et les procédures d'information.
- La pharmacovigilance étant abordée à différents titres et développements au présent rapport, on ne développera pas ici ce domaine important aux facettes multiples qui met en jeu d'autres intervenants.
- La recherche appliquée que mène l'AFSSAPS particulièrement dans quatre domaines (thérapie génique, thérapie cellulaire, sécurité virale et recherches d'alternatives à l'expérimentation animale) doit être rappelée. Il est à noter que la part prise par la thérapie génique a paru excessive à la mission d'audit qui a également posé cette question dans une moindre mesure pour la thérapie cellulaire.
2.3. Les interrogations sur l'expertise
Element primordial de l'évaluation, l'expertise est réalisée par les commissions d'experts (et comités et groupes spécifiques) siégeant auprès de l'AFSSAPS, les deux commissions dont elle assure le secrétariat jusqu'à l'entrée en vigueur de la loi du 13 août 2004 ainsi que par les trois directions de l'évaluation au sein de l'Agence, principalement la direction de l'évaluation et des médicaments et des produits biologiques (DEMEB) (cf. supra 1 ère partie).
La valeur de l'évaluation réalisée par l'AFSSAPS est attestée clairement au plan international ainsi que les instances d'inspection l'ont signalé, mais les conditions de son organisation ont fait l'objet de critiques de fond et de constatations d'insuffisances. Il convient donc de les rappeler tout en les replaçant en perspective compte tenu du niveau des résultats atteints et des évolutions récentes positives.
2.3.1. Expertise interne et expertise externe
Il a été reproché à l'AFSSAPS, notamment par l'audit des inspections et le rapport d'information de la commission des finances du Sénat (précités) de ne pas savoir établir un équilibre entre l'expertise interne, réalisée au sein de l'Agence elle-même et l'expertise externe faite à travers les commissions d'experts dont les membres sont choisis à partir d'une liste établie par le directeur général de l'Agence (1745 experts nommés en 2003 auprès de l'ensemble de ces instances). Il n'y aurait pas de véritable stratégie qui expliquerait cette option.
L'accent mis sur l'expertise externe n'est pas niable ; ainsi le directeur général précise-t-il en octobre 2001 : « Depuis la création de l'Agence, un choix a été effectué dans le cadre de l'expertise française en produits de santé avec le recours à une expertise externe » (cité par l'audit page 26). La tonalité donnée par le tout récent rapport d'activité de l'AFSSAPS pour 2003 (page 50) est un peu différente : « En complément de l'expertise interne qu'elle conduit, l'AFSSAPS fait appel à un réseau d'experts externes placés dans les commissions qui siégent auprès d'elle ». Quel que soit l'accent mis sur cet aspect essentiel de l'organisation de l'évaluation, ce sont les réalités et d'abord les possibilités qui doivent être appréciées concrètement.
La référence à deux pays où les systèmes d'évaluation du médicament reposent principalement sur l'évolution interne est régulièrement avancé : il s'agit de l'Allemagne et des Etats-Unis.
* Caractéristiques des deux catégories d'évaluation
Chacune des deux catégories d'expertise présente ses caractéristiques et donc avantages et inconvénients, mais de ce fait on relève aussi entre elle une complémentarité.
L'expertise interne peut être effectuée par des médecins et des pharmaciens, des biologistes, des biostatisticiens employés par l'AFSSAPS. Cette expertise a l'intérêt d'un certain « professionnalisme » et peut aboutir au maintien de la cohérence et donc d'une homogénéité de l'évaluation d'un dossier à un autre.
Ces professionnels de l'évaluation peuvent cependant avoir l'inconvénient de se couper, avec le temps, de l'actualité de la médecine et surtout de la notion de contact avec les patients auxquels les médicaments sont « in fine » destinés.
Les experts externes ne sont pas des professionnels de l'évaluation. Ils peuvent avoir du mal à dégager le temps nécessaire à l'examen précis des dossiers. Ils ont la particularité de rester en contact constant avec les réalités de l'actualité professionnelle du médecin, du pharmacien, du toxicologue, du biologiste et donc d'être parfaitement sensibles aux réactions potentielles des patients face à un nouveau médicament.
De plus, la possibilité de faire appel à des experts divers fait qu'il est toujours possible, lorsqu'un médicament est destiné à un « nouveau champ thérapeutique » de faire appel à un spécialiste de maladies antérieurement mal connues des professionnels de l'évaluation. Ce peut être le cas pour les maladies rares ou orphelines.
L'équilibre optimal à atteindre entre ces deux expertises est évident au plan théorique.
Dans une première phase, les experts internes devraient étudier tout le dossier, mettre en évidence les études pharmaceutiques, pharmaco-toxicologiques et cliniques essentielles, décrire ces études, donner leurs résultats et, si nécessaire, contrôler, voire préciser, l'obtention de ces résultats (par exemple contrôler les calculs statistiques).
Dans une seconde phase, les experts externes qui sont en contact avec l'actualité médicale et les patients devraient évaluer la signification de ces résultats et, par exemple, lors d'un essai clinique, donner leur avis irremplaçable sur :
la population étudiée : les patients inclus dans les essais sont-ils proches des patients à traiter en France ?
le critère d'évaluation a-t-il une signification médicale forte ?
la différence observée en deux groupes de patients : cette différence sur le critère d'évaluation correspond-elle à un bénéfice médical évident ?
En fonction de ces données et des données de la toxicologie, il sera plus aisé de définir le rapport efficacité/sécurité du nouveau médicament.
- Cet équilibre optimal théorique entre l'évaluation interne et l'évaluation externe suppose une forte évaluation interne, c'est-à-dire un nombre suffisant d'évaluateurs ayant une formation initiale forte et capables d'examiner les centaines de dossiers présentés chaque année (soit des millions de pages).
En pratique, compte tenu de la masse de dossiers à étudier, il est fait appel en partie à l'expertise externe dès la première phase d'évaluation.
* L'audit des inspections générales (page 27) a lui-même indiqué les motivations concrètes qui peuvent expliquer la prédominance donnée à l'expertise externe et les avantages substantiels de cette orientation :
« Les motivations de ce choix sont de plusieurs ordres :
- compenser la faiblesse numérique des moyens internes
- mobiliser les spécialistes de la question sur des dossiers très pointus
- utiliser dans des conditions financières avantageuses une ressource scientifique de haut niveau
La mission estime que ce choix organisationnel présente des avantages, dans la mesure où il permet de disposer d'une expertise scientifique de très bonne qualité. En effet, le fait que les experts auxquels il est fait appel soient en contact direct avec la recherche universitaire ou les laboratoires de recherche des industries pharmaceutiques leur donne une bonne connaissance des derniers développements scientifiques. Par ailleurs, ce système permet de faire appel aux spécialistes nationaux des questions traitées, ce qui est une garantie supplémentaire en termes de sécurité sanitaire.
La mission souhaite, toutefois, que la question du « juste équilibre » entre le recours à l'expertise externe et l'utilisation des ressources internes fasse l'objet d'une véritable réflexion de l'Agence en liaison avec le ministère de la santé ».
La notion de juste équilibre entre le recours à l'expertise externe et l'utilisation des ressources internes ne peut, semble-t-il, constituer une vraie question qu'à l'intérieur d'une marge de choix assez étroitement délimitée par la réalité concrète : ampleur des moyens mis en oeuvre (humains notamment) que l'on peut consacrer à l'expertise interne, valeur réelle des qualités que l'on prête à chacune des deux expertises au regard de la transparence, de l'indépendance, de la motivation, la compétence scientifique étant supposée dans tous les cas.
C'est précisément sur ces aspects que la valeur de chacune des deux expertises doit aussi être appréciée.
2.3.2. Les qualités de l'organisation de l'expertise externe : des progrès à confirmer
La transparence dans la désignation des experts et leur indépendance par rapport à des situations suivies de conflits d'intérêt constituent les défis essentiels.
Les constatations faites fin 2002 par les inspections générales dans leur audit (p. 27) étaient sur ce point critiques :
« Le choix d'un appel massif à l'expertise externe n'est crédible qu'à condition que l'Agence mette au point des modalités transparentes de désignation des experts, un système permettant de garantir leur indépendance et une rémunération de leur travaux qui ne dissuade pas les meilleurs de les effectuer. Or, actuellement ces conditions ne sont pas remplies.
La commission d'AMM peut faire appel à des rapporteurs et des experts choisis sur une liste établie par le Directeur Général de l'Agence.
Les modalités de constitution de cette liste sont insuffisamment explicitées. La transparence du processus de sélection est pour le moins insuffisante puisque aucun appel à candidature ou avis sur les besoins d'expertise n'est publié en amont de la sélection et que la décision n'est pas prise de façon collégiale, mais par les chefs d'unités et de départements de la DEMEB. Ce constat est valable pour d'autres commissions de l'Agence.
- L'absence de garanties suffisantes de l'indépendance des experts.
La garantie de l'indépendance des experts repose essentiellement sur la notion de conflits d'intérêts que l'Agence doit pouvoir détecter quand ils existent. Actuellement, le dispositif mis en place présente de nombreuses lacunes.
- L'Agence n'a pas finalisé de doctrine en matière de conflits d'intérêts.
A partir de la notion de conflit d'intérêts définie réglementairement, l'Agence a établi une classification précise des différents intérêts, autour de laquelle est structurée la déclaration d'intérêts. Cette démarche doit être portée à son crédit ».
Mais on relevait également que la période de référence variait selon les directions concernées : deux ou cinq dernières selon les cas.
Le caractère non exhaustif de la base de données FIDES (fichier informatique des déclarations d'intérêts), son mode de gestion par la cellule de veille déontologique, l'ancrage de celle-ci dans l'organigramme étant lui-même nettement contesté, ont également fait l'objet d'une analyse sévère (p 28) :
« L'Agence ne dispose pas de système global de gestion des experts externes. Le fichier FIDES est géré par la cellule de veille déontologique, en totale déconnexion avec le service des ressources humaines et les directions opérationnelles. Il est destiné à recenser les intérêts déclarés, mais ne peut nullement suivre de manière dynamique le recours aux experts, leur nombre, leur évolution dans le temps.
Par ailleurs, l'objectif principal du fichier qui vise à prévenir la survenue des conflits passe par un enregistrement exhaustif des déclarations d'intérêts des experts. Or, cet objectif n'est pas atteint, faute de procédure de contrôle par recoupement avec d'autres fichiers et d'accessibilité du fichier aux secrétaires des commissions.
La mission a constaté tout d'abord que certains experts ne sont pas enregistrés.
Parmi les experts enregistrés, tous n'ont pas rempli de déclaration d'intérêt. A la demande de la mission, la cellule de veille déontologique a chiffré à 16,5 % en 2000 et 10,5 % en 2001 la part des experts non déclarants des seules commissions dont les déclarations sont publiées par l'Agence.
S'agissant des experts sollicités dans le cadre de l'instruction des dossiers d'AMM, cette part atteint 48 %. De même, sur un échantillon de six groupes de travail dépendant de la commission d'AMM et étudiés par la mission, le taux de déclarations non saisies dans la base FIDES oscille entre 18 et 53 %.
Les déclarations d'intérêts existantes ne sont pas toujours exploitées pour détecter les éventuels conflits d'intérêts ».
Les réponses faites par l'AFSSAPS au cours du premier semestre 2003 dans le cadre de la procédure d'audit traduisent une réelle réaction de remise en ordre, au-delà des orientations déjà affichées :
« Sur la base des travaux déjà engagés par l'Agence, précisément par la cellule de veille déontologique, différentes actions sont en cours et vont permettre, dès cette année, à l'Agence de répondre concrètement aux observations de la mission, en particulier concernant la prévention et la gestion des conflits d'intérêts et la rémunération des experts.
En premier lieu, une révision des listes des experts membres des groupes de travail ou des experts rapporteurs dans les commissions a été réalisée par les directions opérationnelles. Ces listes sont en cours de publication.
Parallèlement, un travail de vérification de la complétude de la base FIDES a été engagé et doit être achevé pour la fin du mois de juin 2003. Ce travail est grandement facilité par la mise en service de la consultation de la base FIDES via Intranet par les secrétariats des commissions (projet FIDWEB, avis favorable de la CNIL reçu à l'AFSSAPS fin mai 2003). Cette consultation par les secrétariats facilitera au quotidien la gestion des conflits éventuels lors des réunions des commissions et des groupes de travail. Par ailleurs, il est prévu de rappeler les règles déontologiques en vigueur lors du renouvellement des commissions. Ces règles sont d'ailleurs précisées dans les appels à candidature diffusés via le site Internet de l'Agence (matériovigilance, publicité, cosmétovigilance). Cette procédure sera progressivement généralisée dans l'année, notamment pour recruter de nouveaux experts rapporteurs.
Le transfert de la gestion des experts externes de la cellule de veille déontologique au département des ressources humaines a été décidé. Il sera effectif à l'automne 2003 ».
La question des rémunérations des experts a également été soulevée. La faiblesse de celles-ci lorsqu'elles existent (médicaments) et leur inexistance dans d'autres domaines (thérapie cellulaire, thérapie génique) ne sont pas de nature à permettre le recrutement des meilleurs parmi un nombre de candidats qui risque de se restreindre. Là aussi, toutefois, des améliorations ont été acquises depuis deux ans.
Enfin, l'urgence d'établir un haut niveau de crédibilité de l'expertise externe par la transparence, l'indépendance et un statut convenable des experts, y compris la rémunération, n'exclut pas de porter son attention aux conditions de l'expertise interne qui a nécessairement moins focalisé l'attention, mais qui n'est pas à l'abri de tout problème.
* En conclusion, on peut considérer comme positives les mesures qui ont été prises pour apporter la clarté et la rigueur indispensable à l'expertise externe qui, d'après les constats argumentés des corps de contrôle, en avait particulièrement besoin. L'effort ne doit toutefois pas être relâché car la tâche de remise à niveau n'est pas achevée et surtout il s'agit d'un domaine dans lequel la vigilance est une exigence permanente. Les situations et donc les dérives potentielles évoluent très rapidement ainsi que le montrent les constatations inquiétantes auxquelles sont arrivées tout récemment les autorités américaines au sujet de la situation déontologique au sein des NIH (National Institutes of Health) notamment. Cela contribuera d'ailleurs à relativiser l'importance de la question de l'équilibre entre l'expertise interne et l'expertise externe au sein de l'AFSSAPS d'autant que c'est précisément dans l'un des pays souvent cités comme référence, les Etats-Unis, que l'expertise de la FDA a été mise en cause avec la crise du Vioxx depuis septembre 2004, après celles en 2001 de la cérivastatine et du traitement hormonal substitutif en 2000.
Les révélations successives et spectaculaires des dernières semaines de l'année 2004 illustrent la nécessité d'une « expertise radicalement revisitée » ainsi que cela est précisé dans les recommandations concluant le présent rapport.
Les difficultés de la montée en puissance telles qu'elles viennent d'être analysées et remises en perspective, notamment par rapport aux plus récentes évolutions, ont révélé des faiblesses du pilotage quotidien comme stratégique de l'AFSSAPS. Cette situation est d'autant plus paradoxale que la qualité de l'expertise française dans le domaine des produits de santé et le niveau de sécurité sanitaire font l'objet de l'avis général et au plan international, d'appréciations très positives. La structure et les mécanismes institués par la loi du 1 er juillet 1998 ne sont pas non plus en cause dans les difficultés enregistrées ; éventuellement il peut s'agir de « legs » antérieurs à 1998 qu'il convient, le cas échéant, de « toiletter ». On estime parvenir à une stabilisation qui serait satisfaisante et même prometteuse si des éléments nouveaux d'origines diverses ne risquaient pas de la remettre en cause.
III. UNE STABILISATION REMISE EN CAUSE
La stabilisation progressive de l'AFSSAPS à un régime de croisière exigeant constitue un objectif à la fois essentiel au regard des impératifs de sécurité sanitaire et ambitieux par rapport aux « rigueurs du terrain » du domaine et aux mouvements institutionnels qui l'affectent.
Les « rigueurs du terrain » dans le domaine des produits de santé illustreront d'abord les multiples développements qui accroissent sensiblement la charge d'activité de l'AFSSAPS ; la dimension européenne des compétences de l'Agence en est souvent à l'origine.
Les mouvements administratifs et institutionnels recouvrent plus précisément les modifications imprévues, voire intempestives, du périmètre de compétences de l'Agence et qui accroissent ses charges.
3.1 La croissance des activités dues aux circonstances
3.1.1. L'exemple de la veille sanitaire
* La fonction de veille sanitaire est exercée par l'AFSSAPS sur la sécurité d'emploi et la qualité des produits de santé. Cette fonction fait appel à la vigilance exercée sur chaque produit, mais aussi au contrôle de la qualité des produits.
Le département des alertes chargé de la gestion de celles-ci propose au directeur général toute mesure de retrait de lots ou de produits et veille à l'exécution des décisions. L'année 2003 a été de nouveau marquée par une forte croissance du nombre de signalements ainsi que l'illustre le tableau ci-dessous :
Veille sanitaire des médicaments
|
Veille accidents pour le médicament |
1997 |
1998 |
1999 |
2000 |
2001 |
2002 |
2003 |
|
Nombre de signalements |
162 |
149 |
182 |
254 |
353 |
424 |
579 |
|
Nombre de décisions de rappels de lots |
60 |
37 |
36 |
47 |
47 |
58 |
64* |
|
Nombre d'alertes sans retrait |
14 |
4* |
*sur les 68 alertes, dont 64 retraits :
- 24 étaient destinées aux officinaux (n° d'alerte en A)
- 9 étaient destinées aux établissements de santé (n° d'alerte en B)
- 13 étaient destinées aux officinaux et aux établissements de santé (n° d'alerte en A/B)
- 22 étaient sans n d'alerte (nombre réduit des destinataires hospitaliers prévenus directement par l'industriel sans passer par le système I-Media avec les fichiers complets).
Le doublement en trois ans du nombre de signalements permet de mesurer la croissance de l'activité de veille.
* La même constatation pouvait être faite pour les enquêtes spéciales. Regroupée au sein d'une équipe dédiée, l'activité des enquêtes spéciales relève du traitement et du suivi administratif, disciplinaire et pénal des affaires relatives à :
des produits de santé mis sur le marché en infraction avec le code de la santé publique et susceptibles de présenter un danger pour la santé publique ;
des entreprises ayant une activité dans le domaine des produits de santé en infraction avec la réglementation.
En 2003, 149 nouveaux dossiers ont été enregistrés auxquels il convient d'ajouter la poursuite de l'instruction des dossiers enregistrés en 2002 (60).
* La veille toxicologique constitue une branche de la coordination des vigilances des produits de santé ; il s'agit ici de la surveillance des effets toxiques des substances chimiques et biologiques (matière première, principe actif, excipient, emballage ...) entrant dans la composition ou la fabrication des produits de santé (médicaments, cosmétiques, dispositifs médicaux ...) et de l'analyse de ces effets pour mener des actions d'alerte, de prévention, de formation et d'information (sécurité sanitaire).
* Par ailleurs, dans le cadre des impératifs de la lutte contre le terrorisme, l'AFSSAPS est amenée à participer à des travaux et recherches évidemment non envisagés par la loi de 1998.
Ainsi, l'Agence participe (sous l'égide de la DGS et du haut fonctionnaire de défense) aux groupes de travail (Biotox et Piratox menace chimique pour ce dernier). Par ailleurs, l'AFSSAPS est chargée du contrôle de l'usage (mise en oeuvre, transport, importation, exportation) de microorganismes ou toxines susceptibles, par utilisation frauduleuse, de porter atteinte à la santé publique. Elle est compétente pour proposer et mettre en oeuvre la réglementation dans ces domaines. Elle participe à l'expertise auprès du ministère des affaires étrangères sur les questions de chimie et de biologie intéressant la défense : contrôle de biosécurité et des agents pathogènes. Enfin, elle est appelée à donner son avis sur les exportations à usage dual.
Ainsi, au-delà des fonctions de veille classique dont le volume peut sérieusement croître en intensité, surtout lorsque certains risques émergents se précisent (cf. infra), l'AFSSAPS est appelée à exercer des fonctions nouvelles de défense dans le cadre d'un périmètre d'activités qui est réputé rester « constant ».
3.1.2. Le nouveau régime des essais cliniques
Les essais cliniques étaient depuis la loi du 20 décembre 1988 régentés par la loi sur la protection des patients, dite « loi Huriet ». La France a acquis dans ce domaine une position de référence et les différents intervenants ont souhaité qu'elle puisse la maintenir. Or certains Etats-membres réduisent les délais et limitent les contraintes (Grande-Bretagne, Allemagne, Belgique). Ainsi, dès 2003 on a enregistré un tassement de 5 % à 10 % des essais déclarés en France contre une croissance du même ordre en Grande-Bretagne et en Allemagne, mais beaucoup plus élevée notamment en Belgique, en Espagne et aux Pays-Bas. L'attractivité de notre pays est, de ce fait, affaiblie.
Une première série de modifications importantes avait été apportée avec la loi du 4 mars 2002 sur les droits des malades : information des participants sur les résultats globaux de la recherche, rôle de la « personne de confiance » dans la recherche en situation d'urgence, dispense d'autorisation préalable pour certaines recherches sans bénéfice individuel direct.
La directive 2001/20/CE implique une révision d'ensemble du dispositif existant jusque là. La transposition de la directive en droit français a été réalisée dans le cadre de la loi de santé publique du 8 août 2004. Le changement fondamental réside dans le passage d'un régime déclaratif simple à un régime d'autorisation des essais. On peut noter d'autres modifications comme l'adaptation des conditions de participation des personnes vulnérables et la mise en place d'une base de données des essais cliniques notamment dans une perspective européenne.
Dans la perspective de ce changement de système, les inspections générales (audit page 33) notaient à la fin de l'année 2002 :
« Ainsi, en matière d'essais cliniques, l'entrée en vigueur au 1 er mai 2004 d'une nouvelle directive va se traduire par une évolution significative de la nature des travaux et leur ampleur. Or, ce surcroît de travail n'a pas été anticipé par le recrutement progressif de personnel scientifique compétent pour évaluer les protocoles d'essais dans des délais contraints, alors même que les missions actuelles de l'unité « essais cliniques » de la DEMEB ne sont que difficilement remplies avec les effectifs actuels. Ce nouveau dispositif affectera également la DIE, s'agissant de l'autorisation parallèle des sites d'essais cliniques ».
La charge accrue de travail pour l'AFSSAPS n'a pas été ignorée. La demande d'autorisation d'essai clinique, qui concerne tous les produits de santé, devra être examinée dans un délai maximal de 60 jours ; au cours de l'essai, l'Agence aura également à se prononcer sur chaque autorisation d'amendement. Enfin, elle aura à réceptionner les informations sur les fins d'essais cliniques et leurs résultats alors qu'aujourd'hui elle ne reçoit que les déclarations de fins d'essais prématurés.
En 2003, à titre d'indication, l'AFSSAPS a enregistré 1100 déclarations d'essais cliniques portant sur les médicaments et 2300 amendements.
Le rapport annuel 2003 de l'AFSSAPS (page 76) précise à ce sujet les conséquences pour elle du nouveau système :
« La mise en oeuvre de la directive nécessite la mise en place de structures à l'AFSSAPS en charge de l'évaluation des essais en France et de leur inspection. La modification essentielle du système actuel provient en effet de l'introduction d'un système d'autorisation des essais - soit implicite, soit explicite - par l'autorité compétente, qui confère à l'AFSSAPS des responsabilités et des contraintes, notamment de délai, nouvelles.
Une phase pilote - Afin de préparer l'ensemble des acteurs du système, l'AFSSAPS propose aux promoteurs depuis novembre 2003 une phase pilote simulant la procédure à venir d'autorisation préalable d'essai clinique. Cette phase se limite dans un premier temps à l'évaluation des dossiers de demande d'autorisation pour essais cliniques de médicaments en phase 1 en France et s'adresse à tous types de promoteurs d'essais. L'objectif est de vérifier si le contenu des dossiers à soumettre est adapté et applicable aux différentes situations et de mesurer la praticabilité du système d'évaluation dans un délai maximum de 30 jours pour ce type d'essais. Elle devrait être étendue dans un second temps à d'autres essais puis au suivi des essais après leur mise en place ».
Il s'agit là encore d'une charge nouvelle qui n'était pas prévue dans les activités déterminées par la loi de 1998.
3.1.3. Le contrôle des vaccins
Depuis le 1993, la France a vu l'activité de contrôle des vaccins en laboratoire augmenter considérablement : libération des lots de vaccins pour laquelle elle est devenue la première autorité nationale pour le nombre. Le rapport annuel 2003 de l'AFSSAPS (page 25) souligne encore une croissance de l'activité et esquisse les conséquences de la nouvelle réglementation :
« Le contexte d'élargissement de l'Union européenne et le fait que la France apporte un fort soutien aux actions de l'OMS auront pour conséquence une demande accrue d'expertise de la part des fabricants de vaccins et des instances européennes et internationales.
Une optimisation des procédures de contrôles et de mise à disposition des vaccins devient nécessaire pour l'AFSSAPS pour, d'une part garantir la qualité des médicaments immunologiques dans des délais compatibles avec les besoins du marché, et d'autre part, pour faire face à des urgences de santé publique (épidémies, contrefaçons), ou apporter un support technique ou réglementaire aux pays tiers.
Une projection basée sur les données conjointes des fabricants et des agences des Nations Unies en charge des programmes de vaccination peut être faite pour les deux années à venir :
Ce nouveau défi européen et international nécessite pour l'AFSSAPS d'ajuster sa stratégie globale de contrôle des vaccins et de poursuivre ses efforts d'investissements en matériels de haute technologie. L'Agence s'y est progressivement préparée par des aménagements de structure sur le site de Lyon, l'appel à l'expertise en matière de bio-statistiques, la mise en place de plans annuels, la réduction des essais in vivo.
Toutefois, cette stratégie de maîtrise des contrôles atteint ses limites dans la mesure où les perspectives nationales, européennes et internationales laissent envisager une extension de la « certification » des vaccins du marché. Une réflexion sur l'évolution de la réglementation européenne à partir des données issues du suivi statistique de la qualité des lots et des observations de non-conformités (1 à 5 % des lots testés suivant la nature du vaccin) devra certainement être initiée ».
3.1.4. La mobilisation due à la dimension européenne
A l'activité de l'AFSSAPS dans la sphère européenne constituée par les essais cliniques, les produits immunologiques (vaccins) et les procédures d'AMM centralisées ou de reconnaissance mutuelle s'ajoutent d'autres domaines.
Il convient de rappeler que dans le cas des procédures d'AMM, la charge de travail accrue par le dispositif européen a pour contrepartie logique une diminution de la charge afférente aux procédures nationales. Mais cette compensation ne s'observe pas dans les autres domaines.
* Ainsi, en matière de contrôle des laboratoires et des sites de fabrication de matières premières à usage pharmaceutique, l'AFSSAPS participe, entre autres, au réseau européen des laboratoires de contrôle des médicaments crée en 1995 par la Direction européenne de la qualité du médicament (dans le cadre du Conseil de l'Europe). Pour le contrôle des médicaments sous AMM européenne centralisée, les laboratoires de l'AFSSAPS sont compétents pour intervenir ; signalons que la France est après l'Allemagne le deuxième pays le plus choisi pour effectuer ces contrôles.
Par ailleurs, l'élaboration de la pharmacopée, actuellement à 81 % européenne, appelle une contribution permanente de la Direction des laboratoires et des contrôles de l'Agence.
* Enfin, les services d'inspection de l'AFSSAPS sont toujours sollicités au sein de l'Union européenne pour des audits, soit dans le cadre de l'élargissement, soit dans celui des procédures de reconnaissance mutuelle avec les pays tiers.
* La participation à l'élaboration des textes européens auxquels peuvent s'ajouter les dispositions nationales constitue une part importante de l'activité de toutes les directions de l'AFSSAPS. On peut citer, outre les questions déjà abordées : les directives relatives aux cellules, aux plantes, à la libéralisation de la publicité vers le grand public pour certains produits, la rédaction de référentiels européens sur les dispositifs médicaux.
* La participation aux nombreux groupes de travail européens mobilisant aussi dans une proportion significative l'énergie des agents de l'AFSSAPS (sur les antibiotiques par exemple).
L'importance de ces charges n'est pas contestée, mais les inspections générales dans leur audit ont mis en lumière la « difficulté qu'éprouve l'AFSSAPS à mesurer la portée de sa mobilisation en matière européenne » (page 31).
Elles ajoutent à ce sujet :
« Même si la mission a bien conscience que tous les aléas du calendrier européen ne peuvent être maîtrisés par l'Agence, elle regrette, néanmoins, qu'aucun travail n'ait pu être conduit avec la tutelle sur ces questions, notamment pour être mieux en mesure d'anticiper les conséquences de la transposition de certaines directives, ou l'application de nouveaux mécanismes européens.
Cette anticipation est rendue plus difficile par le fait que l'Agence n'est pas en mesure de quantifier son actuelle charge de travail induite par l'activité européenne ».
La réponse apportée par l'AFSSAPS à cette remarque illustre la difficulté de cet exercice de mesure :
« Le directeur général prend acte de la nécessité de mieux évaluer l'activité européenne et de travailler à la mesure effective de ces activités. Il souligne cependant la difficulté à bien apprécier, de façon spécifique, l'activité européenne tant elle est, dans les faits, intégrée à l'activité quotidienne. C'est en particulier le cas, comme le souligne la mission, du champ des dispositifs médicaux ou de la pharmacopée, mais aussi de la pharmacovigilance, quelle que soit d'ailleurs la nature des procédures d'AMM (centralisée, de reconnaissance mutuelle ou nationale), ou de certains domaines d'inspection (matières premières en particulier).
Concernant l'harmonisation effective des systèmes de sécurité sanitaires européens, la mission souligne à juste titre l'importance stratégique de cet enjeu. Il est à préciser que la sécurité collective de l'ensemble des citoyens européens est non seulement dépendante de « la capacité collective des Etats membres à y apporter des garanties », mais également de la capacité individuelle de chaque Etat ».
Quant à l'évocation de « l'absence de travail en coordination avec la tutelle », elle a donné lieu à un échange assez vif qui témoigne de certaines difficultés relationnelles avec la DGS. Le directeur général de la santé a en effet noté que :
« L'Agence a toujours cherché à disposer de la plus grande indépendance possible au niveau de sa participation à l'activité communautaire, en invoquant à tort le caractère technique des questions traitées par certaines instances pour obtenir d'y représenter la France seule et en ne respectant pas toujours les règles fixées par le SGCI qui supposent que tous les documents destinés à la représentation permanente et reflétant des positions officielles passent par la DGS pour validation. Dans certains secteurs, la pratique récente a néanmoins montré des améliorations. Mais les modalités de la collaboration indispensable restent à parfaire dans un contexte difficile pour tout le monde qu'entraînent les délais anormalement brefs imposés par les procédures européennes ». (lettre en réponse du 24 juin 2003 déjà citée).
En conclusion de cette analyse des activités européennes de l'Agence, la mission d'audit des inspections générales conclut (page 34) :
« L'émergence d'un système européen avec les risques vus plus haut d'une sécurité sanitaire a minima a justifié jusqu'ici la forte implication de l'Agence à un niveau à la fois technique et stratégique afin d'obtenir des garanties de haut niveau appliquées également à l'ensemble des pays de l'Union.
Si ce souci ne peut être que loué, il n'en demeure pas moins qu'une participation systématique à la totalité des actions européennes, quelle que soit leur implication stratégique ne peut être envisagée à moyens humains constants, les thèmes de travail européens étant amenés à se développer. Aussi, cet enjeu devra-t-il être pris en compte dans le travail de priorisation des missions de l'Agence, à effectuer en liaison avec sa tutelle ».
Cette observation pleinement justifiée sur les possibilités de fonctionnement convenable à moyens humains constants peut être étendue à « l'accroissement des activités autres que de source européenne ». C'est encore plus vrai si l'on tient compte des modifications du périmètre de compétences.
3.2. La modification permanente du périmètre de compétences ; la commission de la transparence
Alors même que toutes les mesures permettant à l'AFSSAPS d'atteindre son régime de croisière dans les domaines nouveaux n'avaient pas été prises, des compétences supplémentaires lui ont été conférées. Il s'agit du FOPIM, puis du secrétariat de la commission de la transparence.
3.2.1. Le FOPIM
* La gestion du FOPIM (Fonds de promotion de l'information médicale et médico-économique) a été confiée par la loi de financement de la sécurité sociale de 2001 à l'AFSSAPS. Ce fonds, qui a été mis en place en 2002, participe totalement ou partiellement aux actions d'information et de communication en matière de bon usage des produits de santé, de prescription et de dispensation des médicaments.
La création de cet organisme semble avoir eu notamment pour objectif de structurer des actions en résolvant les conflits de compétences existant entre la DGS et l'AFSSAPS. Il n'apparaît pas que ce but ait été atteint, ni que la nouvelle structure ait comblé les manques que l'on aurait constatés. Mais les besoins de coordination demeurent et la suppression récente du FOPIM ne suffit pas à y répondre.
* L'éparpillement des fonctions dans le domaine de l'information sur le médicament est délicate ; elle a entraîné un enchevêtrement des fonctions. En présentant les choses simplement, on peut indiquer que jusqu'en 2003, l'investigation est faite par l'ANAES, la vigilance est assurée par l'AFSSAPS, la transparence par la Direction générale de la santé (commission de la transparence), les autres intervenants étant le FOPIM (alors dans la mouvance de l'AFSSAPS), et les caisses d'assurance maladie.
L'audit des inspections générales a souligné la difficulté d'organiser des missions qui se situent à la frontière des compétences de chacun notamment pour l'AFSSAPS, et donnait des esquisses (page 57) de solution :
« L'ensemble des activités qui s'appuie sur l'évaluation scientifique relève du champ de compétence de l'Agence. Ainsi, aucune ambiguïté n'existe s'agissant des vigilances.
En ce qui concerne l'observation des conditions réelles d'utilisation des produits de santé, il s'agit bien d'une mission de l'Agence, mais il est recommandé de supprimer l'observatoire des prescriptions et de réintégrer ses compétences dans un service propre de l'AFSSAPS.
Cette réintégration suppose une réflexion sur la finalité de l'évaluation postérieure à la mise sur le marché et sur les modalités d'organisation de l'Agence pour y répondre. En l'espèce, au vu des acteurs compétents en la matière dans le paysage institutionnel français, la structuration d'un département en charge de la maîtrise d'ouvrage de ces études, c'est-à-dire capable de piloter des études pharmaco-épidémiologiques sans les réaliser, constitue une voie médiane préconisée par la mission (...).
Par ailleurs, il convient de renforcer les travaux liés à la production de référentiels de bonne pratique, mission qui relève pleinement de l'Agence.
Cette action nécessite la constitution d'un pôle unique en charge de la production des référentiels de bonne pratique (fiches de transparence, recommandations de bonne pratique, fiches d'information thérapeutique). Ce pôle dédié à l'information scientifique trouverait efficacement sa place à la DEMEIS en raison des synergies possibles avec le contrôle de la publicité et l'activité de la commission de la transparence, le cas échéant. Il devrait travailler en étroite collaboration avec le département des vigilances de la DEMEB.
En matière de communication vers les professionnels de santé ou les usagers du système de soins, la frontière entre ce qui relève légitimement de l'Agence et ce qui pourrait être externalisé, est ténue. Si la production de référentiels techniques relève de l'Agence, leur diffusion requiert un réel renforcement de la cellule communication. En revanche, la mission estime que tout ce qui s'inscrit dans une logique de modification du comportement des acteurs ne relève pas de la sécurité sanitaire.
A cet égard, elle considère que l'activité du FOPIM risque de créer une confusion avec la propre communication de l'Agence. Dans ces conditions, la mission préconise qu'une réflexion soit engagée sur la possibilité de détacher cet organisme de l'AFSSAPS ».
3.2.2. La commission de la transparence
Le ministère de la santé a souhaité rationaliser cette organisation et un choix a été exprimé par un décret de septembre 2003 qui a transformé la direction des études médico-économiques et de l'information scientifique (DEMEIS) de l'AFSSAPS en créant un secrétariat général à la commission de la transparence (le titulaire de la DEMEIS a lui-même été nommé à ce nouveau poste) et lui rattachant le FOPIM.
L'un des principes de cette réforme était « d'autonomiser » le secrétariat de la commission de la transparence, conformément à sa spécificité qui la distinguait nettement des autres commissions. Son caractère scientifique exclusif a été affirmé et maîtrisé ; les 20 membres titulaires ayant voix délibérative sont désormais des scientifiques, les huit membres représentant les administrations, les représentants des organismes d'assurance maladie et de l'industrie pharmaceutique siégeant désormais à titre consultatif.
Ce réaménagement des structures répond à un souci de cohérence en mettant fin à une complexité excessive, elle-même génératrice de pertes d'énergie et de frictions ; toutefois, elle contredisait une observation faite par les inspections générales (page 58) :
« Il existe un lien entre l'appréciation du service médical rendu effectué par la commission de la transparence et les compétences médico-scientifiques de l'Agence. Pour cette raison, le rattachement de cette commission à l'Agence est justifié. En revanche, les décisions relatives à l'admission du remboursement ne relèvent pas de l'AFSSAPS ».
On touche la difficulté liée à l'ambivalence de travaux d'analyse scientifique, y compris le rapport bénéfice/risque et l'analyse économique au sens du service médical rendu.
* Cette nouvelle structuration des compétences n'aura pu être vraiment appréciée puisqu'une nouvelle organisation est mise en place avec le transfert de la commission de la transparence au sein de la Haute Autorité de Santé crée par la loi relative à l'assurance maladie du 13 août 2004 et la suppression de l'ANAES intégrée au sein de la nouvelle HAS. L'approche économique a ainsi entraîné l'instance qui a aussi en charge l'appréciation médico scientifique du médicament en dehors du périmètre de l'AFSSAPS.
En outre, la nouvelle HAS accueillera en son sein la CEPP (commission d'évaluation des prestations et des produits).
Le nouvel article L. 161-37 du code de la sécurité sociale (art. 35 de la loi du 13 août 2004) précise ainsi les compétences de la nouvelle HAS :
« Art. L. 161-37 - La Haute autorité de santé, autorité publique indépendante à caractère scientifique dotée de la personnalité morale, est chargée de :
1°) Procéder à l'évaluation périodique du service attendu des produits, actes ou prestations de santé et du service qu'ils rendent, et contribuer par ses avis à l'élaboration des décisions relatives à l'inscription, au remboursement et à la prise en charge par l'assurance maladie des produits, actes ou prestations de santé ainsi qu'aux conditions particulières de prise en charge des soins dispensés aux personnes atteintes d'affections de longue durée. A cet effet, elle émet également un avis sur les conditions de prescription, de réalisation ou d'emploi des actes, produits ou prestations de santé et réalise ou valide des études d'évaluation des technologies de santé.
2°) Elaborer les guides de bon usage des soins ou les recommandations de bonne pratique, procéder à leur diffusion et contribuer à l'information des professionnels de santé et du public dans ces domaines, sans préjudice des mesures prises par l'Agence française de sécurité sanitaire des produits de santé dans le cadre de ses missions de sécurité sanitaire.
3°) Etablir et mettre en oeuvre des procédures d'évaluation des pratiques professionnelles et d'accréditation des professionnels et des équipes médicales mentionnées à l'article L. 1414-3-3 du code de la santé publique ; (...)
En outre, d'autres dispositions précisent les prérogatives de la nouvelle HAS en matière « d'évaluation de service attendu d'un produit, d'un acte ou d'une prestation de santé » à laquelle elle peut procéder, mais prévoit également que :
« Sans préjudice des mesures prises par l'Agence française de sécurité sanitaire des produits de santé dans le cadre de ses missions de sécurité sanitaire et notamment celles prises en application du 2° de l'article L.5311-2 du code de la santé publique, la Haute autorité de santé fixe les orientations en vue de l'élaboration et de la diffusion des recommandations de bonne pratique de l'Agence française de sécurité sanitaire des produits de santé mentionnée à l'article L. 5311-1 du même code et procède à leur diffusion ».
Dans ce domaine des référentiels de bonne pratique, l'AFSSAPS verra donc « ses orientations en vue de l'élaboration et de la diffusion des recommandations de bonne pratique » fixées par la Haute Autorité de Santé, autorité publique indépendante à caractère scientifique tout en continuant à être sous la tutelle de la Direction générale de la santé du ministère.
Un schéma publié le 27 janvier 2005 par le journal « Le Monde » illustre bien le positionnement du « nouvel organisme dans la galaxie du contrôle de la santé ».
Un nouvel organisme dans la galaxie du contrôle de la santé
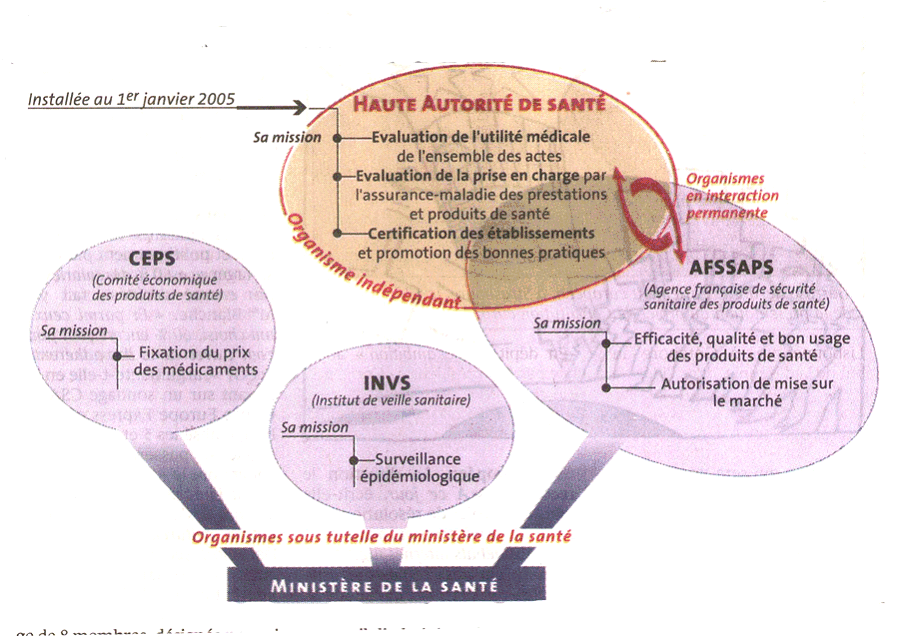
Source : Le Monde du 27 janvier 2005
La simplification souhaitée des différents côtés ne semble pas trouver ici la concrétisation que l'on pouvait espérer. La situation inédite dans laquelle ce domaine va se trouver nécessitera certainement de nouvelles clarifications.
IV. UNE ACTUALITÉ LOURDE D'INTERROGATIONS
Au-delà de l'évaluation du fonctionnement de l'AFSSAPS, ainsi que des mécanismes auxquels elle participe avec d'autres acteurs français et européens, des interrogations se posent dans son « coeur de métier », essentiellement sur la iatrogénie médicamenteuse et les essais cliniques dont l'expertise est dépendante. L'ampleur des problèmes qui rentrent dans ce cadre dépasse largement non seulement l'AFSSAPS elle-même, mais toutes les agences et administrations compétentes en matière de médicaments. Il ne s'agit donc en aucun cas d'une mise en cause ciblée de l'AFSSAPS qui, n'a guère à craindre les comparaisons internationales, mais de la présentation d'une vaste question qui constitue le fondement même de la sécurité des produits de santé, en l'occurrence des médicaments.
Dans le cadre limité de cette évaluation, on rappellera d'abord la nécessité de la pharmacovigilance renforcée par les accidents iatrogènes et on indiquera les inquiétudes pointées par les praticiens. Plusieurs cas de médicaments soulevant de véritables crises au niveau mondial depuis 2000 constitueront ensuite des illustrations de cette problématique difficile.
4.1. La iatrogénie médicamenteuse
Le médicament est ambivalent, mais doit par principe présenter un bilan d'utilisation positif. Ce constat conduit à poser le problème des conséquences néfastes de son usage, « la iatrogénie médicamenteuse ».
La définition du concept doit être précisée. Celle présentée par le rapport de mission ministérielle sur « la iatrogénie médicamenteuse et sa prévention » 34 ( * ) vise les éléments suivants (sachant que la iatrogénie en général est définie par le dictionnaire Robert de la langue française comme « toute pathologie d'origine médicale ») :
« - les effets indésirables sans mauvais usage des thérapeutiques ou aléas non fautifs ;
- les effets indésirables avec mauvais usage des thérapeutiques, que ce « mauvais usage » soit le fait du médecin ou, d'autres soignants ou encore du malade lui-même, par automédication inappropriée ou mauvaise observance du traitement.
Cette conception s'éloigne du sens premier et étymologique, mais il respecte l'esprit de notre démarche : la meilleure connaissance des effets indésirables des thérapeutiques dans le but de rechercher tous les moyens de les prévenir ou à défaut de les limiter ».
4.1.1. Des références américaines
* La question de l'iatrogénie médicamenteuse est identifiée depuis longtemps comme essentielle. Ainsi, une méta-analyse d'études a évalué à 100.000 environ le nombre annuel de décès par effet indésirable médicamenteux aux Etats-Unis chez les malades hospitalisés (J. Lazarou - incidence of adverse drug reactions in hospitalized patients. JAMA 1998.279.
Dans son rapport précité (mars 1998, page 27), le Pr. Patrice Quéneau donne un éclairage de la dimension médico-économique du problème à travers une étude américaine de 1997 qu'il cite dans les termes suivants :
« L'approche médico-économique est difficile et les données épidémiologiques précises peu nombreuses. Cependant, les enjeux financiers sont majeurs. Aux Etats-Unis, un travail récent de D.W. Bates et huit autres chercheurs (The costs of adverse drug events in hospitalized patients JAMA 1997/277) rapporte que le coût annuel de la morbi-mortalité liée aux effets indésirables des médicaments serait de l'ordre de 76,6 milliards de $ US, dont la majorité (47 milliards de $ US) serait due à l'hospitalisation pour accident thérapeutique ou absence de traitement approprié. Ces auteurs soulignent que :
- ce coût dépasse de beaucoup celui du diabète, évalué à 45,2 milliards de $ US.
- Le coût des accidents qu'ils jugent « évitables » (« preventable ») est plus élevé que celui des accidents inévitables. En effet :
- pour l'ensemble des accidents médicamenteux, l'allongement moyen de la durée de l'hospitalisation est de 2,2 jours, et le surcoût total moyen par malade de 3244 $ US.
- pour les accidents médicamenteux jugés « évitables », l'allongement moyen de la durée de l'hospitalisation est de 4,6 jours et le surcoût total moyen par malade de 5857 $ US.
- ce coût élevé justifie à lui seul des efforts de prévention, indépendamment des raisons humanitaires qui les justifient en toute priorité ».
Dans l'article cité ci-dessous, résumant l'étude faite dans les services d'accueil et d'urgence français en 1999, le même auteur, le Pr. Patrice Quéneau, indique :
« Des études récentes de cohorte ont par ailleurs estimé le nombre annuel de décès liés aux complications digestives graves (hémorragies, perforations) des anti-inflammatoires non stéroïdiens à environ 2000 en Grande-Bretagne et 16.500 en Amérique du Nord, chiffre voisin du nombre de décès dus au Sida. Par extrapolation, Tramer émet l'hypothèse d'un décès par complication digestive grave pour 1220 patients traités par AINS pendant deux mois ou plus. En outre, le « surcoût iatrogène » lié aux effets indésirables médicamenteux est considérable ».
* L'ampleur de la iatrogénie médicamenteuse se constate naturellement en dehors des Etats-Unis. Toutefois deux éléments d'inégale importance méritent d'être rappelés.
D'une part la philosophie dans laquelle les procédures d'AMM s'insèrent n'est pas exactement la même qu'en Europe et en France en particulier. L'accent est prioritairement mis pour l'efficacité dans le cadre de l'analyse bénéfice/risque. Ce que l'on observe dans les cas examinés ci-après au sujet de la cérivastatine, du traitement hormonal substitutif et actuellement du Vioxx montre clairement que les préoccupations et limitations d'emploi, les contre-indications impératives et l'exigence d'un réel bénéfice par rapport à d'autres spécialités, ne se recouvrent pas des deux côtés de l'Atlantique.
D'autre part, les conditions de dispensation du médicament sont encore plus différentes entre les Etats-Unis et la France. Le système de distribution particulièrement contrôlé qui est le nôtre à travers le monopole de la vente du médicament en officine a nécessairement des effets importants sur la iatrogénie médicamenteuse. C'est là une donnée essentielle.
4.1.2. Des études françaises
Dans le prolongement du rapport précité du Pr. Patrice Quéneau, deux études permettent de préciser le phénomène en France.
* En mai 1999 a été publiée l'étude engagée au printemps 1997 sur « les hospitalisations dues à un effet indésirable médicamenteux », et fondée sur une enquête réalisée auprès d'un échantillon représentatif des services de spécialités médicales des hôpitaux publics français par les Centres régionaux de pharmacovigilance, le rapport final étant rédigé par les Pr. Pierre Pouyanne, Françoise Maramburu, Jean-Louis Imbs. Ce dernier est en outre l'auteur de la présentation donnée dans la revue Thérapie (1999-54) dont sont extraites les citations ci-après :
« Le réseau des Centres régionaux de pharmacovigilance a estimé la prévalence des effets indésirables médicamenteux sur un échantillon représentatif (sondage en grappe stratifié à trois degrés, par tirage au sort) des malades hospitalisés dans des services de médecine, chirurgie et long séjour publics. L'enquête a été effectuée un jour donné, au cours du printemps 1997. Chaque cas d'effet indésirable a fait l'objet d'une validation. L'échantillon d'étude porte sur 2132 malades dont 969 hospitalisés en Centre Hospitalier Universitaire et 1163 en Centre Hospitalier Général. Sa répartition selon les spécialités et les structures d'hospitalisation est représentative de l'ensemble des hospitalisations dans les hôpitaux publics métropolitains. Au moins un effet indésirable validé était présent chez 221 patients le jour de l'enquête, soit un taux de prévalence de 10.3 pour cent (IC pour cent : 26 à 42 pour cent), ces effets étaient graves. A partir d'un taux d'incidence un jour donné de 1.8 pour cent (IC 95 pour cent : 1.0 à 2.5 pour cent), il est possible de calculer que chaque année environ 1.300.000 patients présentent un effet indésirable médicamenteux au cours d'une hospitalisation ».
La conclusion de l'étude elle-même remet en perspective le problème dans les termes suivants :
« Cette étude, la première menée à l'échelon d'un pays, sur un échantillon représentatif de services de médecine et spécialités médicales, met en évidence l'importance du problème des hospitalisations secondaires à la survenue d'un effet indésirable médicamenteux. Le nombre d'hospitalisations (128 768 entrées), le nombre de journées d'hospitalisation (1.146.035), le coût généré par ces hospitalisations (2,1 milliards de francs) montre bien qu'il s'agit d'un problème majeur de santé publique, au même titre que les accidents de la route. Cependant, ces chiffres ne sont le reflet que d'une partie du problème. En effet, ils ne prennent pas en compte tous les aspects de la iatrogénie médicamenteuse, en particulier tous les accidents graves qui ne sont pas hospitalisés notamment les décès, les hospitalisations dans des établissements privés et enfin tous les accidents survenus en cours d'hospitalisation qui vont être à l'origine d'une prolongation d'hospitalisation ou même d'un décès.
* Abordant la question sous un angle un peu différent, celui des signalements par les services d'accueil et d'urgence d'hôpitaux publics français, l'étude réalisée sous la conduite du Pr. Quéneau avec six autres médecins universitaires et l'APNET (Association pédagogique nationale pour l'enseignement de la thérapeutique) a fait l'objet d'une publication dans le Bulletin de l'académie nationale de médecine (2003-187 n° 4 page 647-670 intitulée « Effets indésirables médicamenteux observés dans les services d'accueil et d'urgences françaises ».
« L'importance de la pathologie médicamenteuse comme cause d'hospitalisation est un problème majeur de santé publique. Notre étude a consisté à recueillir systématiquement toutes les observations d'effets indésirables médicamenteux pendant deux semaines (la première en juin 99 et la seconde en décembre 99) dans 10 Services d'Accueil et d'Urgences (SAU) de 10 Centres Hospitaliers : 5 Centres Hospitaliers Universitaires (CHU) et 5 Centres Hospitaliers non universitaires (CH). Sur un total de 1937 patients admis aux 10 SAU pendant ces deux périodes, nous avons retenu les 1562 patients ayant pris au moins 1 médicament au cours de la semaine précédente. Parmi eux, 328 (21 % ; intervalle de confiance à 95 % : 19 %-23 %= avaient consulté en raison d'un effet indésirable médicamenteux (EIM). Au total, 410 médicaments furent incriminés dans la survenue des 328 EIM. Les psychotropes (n = 84 : 20,5 %), les diurétiques (n = 48 : 11,7 %), les anticoagulants (n = 38 : 9,3 %), d'autres médicaments cardiovasculaires (n = 63 : 15,4 %), les antalgiques et les anti-inflammatoires non stéroïdiens (n = 57 : 13,9 %) furent les classes médicamenteuses les plus fréquemment incriminées. Dans 106 cas (37,9 %), l'EIM fut considéré comme évitable en raison d'un mauvais usage du médicament ».
4.2. Quelques exemples de crises récentes
Les accidents ou les incidents graves enregistrés à une large échelle à partir de médicaments sont sans doute aussi anciens que les médicaments eux-mêmes. La pharmacovigilance a précisément permis qu'ils soient mieux et plus vite repérés. Il convient donc de rappeler d'abord cette évidence souvent oubliée pour éviter que se répandent des idées fausses de type : « il y a de plus en plus d'accidents avec les médicaments » ou « les médicaments sont de plus en plus dangereux ». Le bismuth, médicament retiré depuis près de trente ans après avoir été utilisé par des millions de personnes pendant près d'un siècle en est un bon exemple. Sa dangerosité, constatée lors de son retrait dans la forme galénique sous laquelle il était alors vendu, demeure indiscutable, illustrant le caractère permanent de cette question.
Trois cas récents dont l'un se développe actuellement sous nos yeux (le Vioxx) peuvent servir d'éléments de référence pour apprécier le fonctionnement de l'AFSSAPS et les mécanismes de sécurité sanitaire au niveau national, européen et mondial.
4.2.1. Le retrait de la cérivastatine
La cérivastatine est une molécule parmi l'ensemble des statines ; celles-ci visent à réduire l'hypercholestérolémie dans le sang et constituent un des progrès médicaux les plus substantiels des quinze dernières années. Les produits de cette classe thérapeutique ont été et sont considérés comme très bien tolérés, le cas de la cérivastatine étant mis à part depuis la révélation d'accidents qui ont entraîné son brusque retrait le 8 août 2001.
La cérivastatine a été mise en cause en raison d'atteintes musculaires (rhabdomyolyse) qui peuvent revêtir un caractère sévère, voire fatal. Cette molécule était produite par les laboratoires Bayer en France sous les noms des spécialités Staltor et Cholstat.
Dans le cadre de la procédure de reconnaissance mutuelle au niveau de l'Union européenne, le Royaume-Uni était l'Etat-membre de référence et l'AMM européenne délivrée en 1997 portait sur des dosages de 0,1 et 0,3 mg ; un dosage de 0,4 mg avait été autorisé en 2001. Il est important de noter qu'un dosage de 0,8 mg avait été autorisé en août 2000, puis retiré le 1 er août 2001, quelques jours avant le retrait mondial.
Plusieurs éléments d'alerte avaient précédemment été enregistrés depuis décembre 1999 en focalisant progressivement les deux facteurs de risque dans l'utilisation de cette molécule : le dosage supérieur à 0,4 mg d'une part, l'association avec le gemfibrozil (hypo-cholestérolémiant de la classe des fibrates, commercialisé en France sous le nom de Lipur) ; la FDA américaine avait décidé de contre-indiquer cette association en décembre 1999 ; la contre-indication a été insérée à la demande de l'Agence britannique du médicament.
En avril 2001, les autorités européennes compétentes signalent à l'Agence britannique des cas de rhabdomyolyse (67 % d'association cérivastatine -gemfibrosil) dont 3 décès.
Le 8 août 2001, Bayer annonce unilatéralement le retrait mondial des spécialités à base de cérivastatine à l'exception du Japon où précisément le gemfibrozil n'était pas commercialisé, donc sans risques d'association. Ce retrait sera toutefois étendu au Japon, la commercialisation de ce dernier devenant prévisible.
Les différentes agences du médicament annoncèrent alors le retrait et le Secrétaire d'Etat à la Santé du gouvernement allemand critiqua vivement l'attitude de Bayer en la qualifiant « d'inacceptable » car l'Agence allemande du médicament n'avait reçu le rapport de pharmacovigilance fait par le laboratoire et daté du 15 juin 2001 que le 8 août et n'avait pas été informée du retrait de la cérivastatine en même temps que l'Agence britannique. D'une façon générale, Bayer s'est vu reprocher des faiblesses face à ses obligations d'information, transmettant tardivement et incomplètement, d'une manière dispersée, des éléments qu'il était seul à détenir dans leur globalité.
Le retrait mondial était justifié par un nombre important de rhabdomyolyses fatales, notamment aux USA. Le laboratoire Bayer annonçait 59 cas de rhabdomyolyses fatales en août 2001, puis 99 cas en octobre 2001. La survenue d'un tel effet indésirable fatal était évidemment incompatible avec le bénéfice potentiel d'un médicament hypocholestérolémiant (6 millions de patients dans le monde).
Il est à noter que sur les 59 cas de rhabdomyolyses graves rapportés en août 2001, la posologie était souvent élevée : 21 cas à 0,8 mg, 1 cas à 0,6 mg. L'association gemfibrozil était retrouvée dans 12 cas.
En France, la cérivastatine était prescrite à environ 500.000 patients et représentait 12 % du marché des statines. Un seul décès, d'ailleurs très mal documenté par le laboratoire Bayer et dont l'imputabilité était incertaine, était connu.
En pratique, il apparaissait à cette époque que les dangers de l'utilisation de la cérivastatine étaient plus importants aux USA et dans d'autres pays européens qu'en France.
Les raisons en sont les suivantes :
- les posologies unitaires du marché français étaient à 0,1 - 0,3 et 0,4 mg. Depuis 2000 aux USA et 2001 au Royaume-Uni, une dose unitaire était proposée à 0,8 mg. Cette dose n'a pas eu d'AMM en France. Les recommandations françaises comportaient une prescription initiale à 0,1 mg par jour et, le cas échéant, une montée progressive en fonction de la cholestérolémie avec une dose maximale de 0,4 mg.
- l'association avec le gemfibrozil : les RMO françaises, depuis de nombreuses années, précisaient qu'il n'y avait pas lieu d'associer statine et fibrate du fait du risque d'addition des effets indésirables. Dans le cas particulier de la cérivastatine, l'association avec le gemfibrozil était contre-indiquée du fait du risque d'effets indésirables graves. Tel n'était pas le cas aux USA et dans certains autres pays.
Il est à noter d'ailleurs qu'une étude réalisée spécialement pendant l'été 2001 par la CNAMTS sur 360.000 ordonnances de cérivastatine a montré que la posologie maximale de 0,4 mg n'était dépassée que dans 1,61 % des cas et qu'il n'y a eu qu'un pourcentage négligeable d'association au gemfibrozil (0,0035 %).
La gestion du dossier de la cérivastatine par l'AFSSAPS appelle des appréciations positives, voire très favorables, plutôt que des critiques qui ne pourraient qu'être marginales dans un paysage mondial où d'autres ont manifestement eu du mal à prendre en temps utile la mesure du problème.
Les experts associés à la mission des quatre inspections générales pour l'évaluation de la loi du 1 er juillet 1998 ont choisi le cas du retrait de la cérivastatine parmi les neuf exemples sur la « sécurité sanitaire en action ». La conclusion (page 100) qu'ils en tirent, citée intégralement ci-après, illustre la nécessité d'actions pour améliorer la pharmacovigilance, en soulignant que celle-ci doit être coordonée au niveau européen et mondial. Les limites de l'exercice sont d'ailleurs évoquées à la fin de ce texte et obligent, en effet, à une certaine modestie si l'on veut rester dans le domaine des propositions réalisables :
« Conclusion :
Le cas du retrait de la cérivastatine illustre l'importance de la diversité des expertises et de la responsabilisation des industriels.
On peut poser deux sortes de jugements d'ensemble sur cette affaire, si l'on part du postulat (que nous ferons) que le retrait de la cérivastatine était scientifiquement justifié et non prématuré pour la sécurité sanitaire étant donné la disponibilité sur le marché de molécules d'efficacité identique et de risque moindre.
Le premier jugement porte sur l'intérêt et l'efficacité de l'expertise interne au laboratoire pharmaceutique qui a su, contre son intérêt immédiat apparent, prendre une décision qui s'est avérée, même si l'on peut contester la mise en oeuvre et le plan de communication l'accompagnant, rapide, nécessaire, efficace et définitive. Doit-on laisser les industriels décider seuls sur ces questions ?
Le second jugement, sans doute plus sévère, concerne la pharmacovigilance et la pharmaco-épidémiologie au niveau international. Ces structures n'ont pas su prendre une décision avant celle de l'industriel, alors qu'elles étaient en possession de (presque) tous les éléments du dossier. Les structures de recueil de la pharmacovigilance ont eu de petites défaillances de part et d'autres et un certain flottement a entouré l'annonce du retrait, notamment sur le nombre exact de décès rapportés dans le monde, mais on peut dire que globalement elles ont bien rempli leur première mission de recueil et d'échange d'informations. Si les personnes en charge de la pharmacovigilance n'ont pas mesuré l'urgence de la gestion du risque, il se pose alors la question de l'accessibilité des bases de données à un plus grand nombre de partenaires, voire à tous les citoyens, comme cela est réalisé aux USA, dans le cadre du « Freedom of Information Act ». Le pays rapporteur de l'AMM d'un produit (le Royaume-Uni dans le cas de la cérivastatine) était désigné comme le pays responsable de la coordination de la vigilance de ce produit une fois commercialisé ; il n'a pas eu la même évaluation que le laboratoire sur l'urgence du risque. Il apparaît clairement qu'il ne disposait pas de l'estimation comparative du risque avec les produits de la classe. Dans la situation particulière de ce produit qui présentait davantage d'effets indésirables graves que les alternatives d'efficacité équivalente, il n'existait pas de procédure pré-établie permettant de balayer systématiquement et rapidement (éventuellement de façon automatisée) les données rapportées afin de détecter un éventuel signal au niveau de la classe. Il n'existait pas non plus de procédure permettant de décider à partir de quand il fallait retirer le produit. Aucune méthode n'a été mise en oeuvre pour permettre de se prononcer sur le caractère tolérable ou non du risque, ni sur le degré d'urgence de la décision qu'il convenait éventuellement de prendre. Il est probable que l'investissement recherche soit largement insuffisant dans ce domaine d'analyse et d'interprétation du signal.
En annonçant par voie de presse le 8 août 2001 le retrait mondial de la cérivastatine, le laboratoire Bayer provoquait la surprise, mais aussi l'inquiétude. Les patients exposés et les prescripteurs, mais aussi les autorités de santé n'avaient pas pu être informés avant le reste du public - pour des raisons qui semblent liées aux lois du marché boursier - ce qui pose évidemment un réel (et peut-être inédit) problème de communication et d'information sur le risque sanitaire dans le domaine du médicament (et bientôt sans doute des dispositifs médicaux). L'industrie du médicament souffre d'un déficit d'image auprès du public, et sait peu tirer profit de son métier et de sa contribution à l'amélioration de l'état de santé de la population. Ce type de crise ne fait qu'aggraver cette image, semblant donner raison à tous les stéréotypes convenus et durablement négatifs : la bourse passe avant la vie, le profit avant la sécurité sanitaire, le secret avant la transparence, la dissimulation avant l'exhaustivité et la clarté des informations transmises, etc ... Or, sans la décision des laboratoires Bayer le 8 août au matin, il n'est pas exclu que la cérivastatine aurait pu rester encore plusieurs semaines, mois ou années sur le marché, exposant de ce fait un nombre encore plus grand de patients au risque de rhabdomyolyse d'évolution parfois fatale. Il manque clairement un arbre décisionnel permettant d'aider la prise de décision lorsqu'un niveau de risque est atteint (niveau absolu, niveau relatif aux autres médicaments de la classe, fraction attribuable du risque). Mais qui est capable de réaliser un tel arbre ? A-t-on jamais fixé un seuil tolérable de risque pour les médicaments dans l'absolu (comme dans le domaine environnemental, où des seuils de 10 -5 ou 10 -10 ont été proposés selon les contextes) ? Enfin, on peut suggérer que le Conseil Scientifique de l'AFSSAPS se saisisse de ce type de dossiers pour en faire plus systématiquement l'analyse du retour d'expérience, ce qui permettrait de tirer les enseignements et les leçons du passé et d'améliorer le système à l'avenir ».
4.2.2. La remise en cause du traitement hormonal substitutif
La publication des résultats d'enquêtes de grande ampleur réalisées principalement aux Etats-Unis et en Grande-Bretagne 35 ( * ) a entraîné des interrogations, puis une remise en cause du caractère presque systématique des troubles de la ménopause et de la prévention de l'ostéoporose. On estimait en effet pour la France, qu'en août 2003, 30 % à 50 % des femmes âgées entre 48 et 64 ans suivaient un THS (traitement hormonal substitutif). D'autres bénéfices avaient été progressivement attribués, un peu vite semble-t-il, à ces traitements. Or, si certains avantages ont été confirmés, la mise en évidence de nouveaux risques de cancer (sein notamment, endomètre) et d'accidents cardiaques et circulatoires ont amené la plupart des autorités sanitaires des pays concernés à réévaluer le rapport bénéfice/risque des THS.
La question se complique du fait que ces études ayant été réalisées hors de France, le caractère transposable des conclusions n'est pas évident car, là encore, les pratiques peuvent varier sensiblement et, en outre, les doutes ont renforcé les préconisations qui différencient la France de la situation américaine :
- plus faible dosage des produits ;
- durée limitée du traitement (le plus souvent trois ans) ; objectif de limitation systématique à l'âge de 60 ans ;
- exclusion systématique de femmes présentant un seul facteur de risque.
Les controverses très vives qui ont eu lieu aux Etats-Unis et en Grande-Bretagne sur ce sujet se retrouvent en France, mais selon des termes différents. Ce qui peut être souligné ici, c'est la position difficile dans laquelle l'AFSSAPS s'est retrouvée. Ainsi, celle-ci a eu à faire face à la publication (Le Monde du 8 février 2003) d'un appel émanant d'une partie des membres d'un de ses comités d'experts qui estimaient « que les recommandations de l'Agence ne reflétaient ni notre opinion, ni la nature des débats et discussions qui se sont déroulés lors d'une réunion multidisciplinaire en octobre 2003. Nous invitons les médecins prescripteurs à consulter les recommandations de l'association française pour l'étude de la ménopause, plus conformes et plus adaptées à la situation actuelle » (cité par le rapport des experts des quatre inspections générales).
Sur une question où les certitudes scientifiques sont en cours de constitution et où, de l'avis de certains, on s'attend à une diminution importante des prescriptions par crainte de poursuites judicaires contre les médecins, on se contentera de citer la conclusion des experts des inspections générales (rapport des experts page 107). Elle pose avec franchise à la fois les données spécifiques de cette question sous l'angle médico-social, et précise les limites ontologiques de la pharmacovigilance, et donc des agences sanitaires :
« Conclusion
Cette affaire pose plus largement le problème de la sécurité sanitaire des produits « politiquement corrects ». Fruits d'une sorte de conquête en partie féministe pour retarder les conséquences de la ménopause, ces produits ont - un peu rapidement et sur la base d'études de faible niveau de preuve - été considérés comme inoffensifs, voire comme ayant des effets protection sur le risque cardio-vasculaire, sur la démence sénile ou sur le cancer du colon. Aujourd'hui, en dehors de l'indication relative aux troubles du climatère, il n'est pas certain que ces produits aient de réels bénéfices, ni n'améliorent la qualité de vie des femmes traitées, mais il est devenu clair qu'ils favorisent le risque de cancer du sein, peut être aussi celui de l'ovaire, vraisemblablement aussi le risque cardio-vasculaire et celui de démence sénile. La question n'est pas tranchée vis-à-vis de la protection contre le cancer colo-rectal.
Cependant ces produits sont toujours sur le marché, sans doute parce qu'il aurait été considéré comme un séisme social de retirer brutalement des produits prescrits chez 30 à 50 % d'une tranche d'âge de la population féminine française. Probablement disparaîtront-ils complètement de la prescription au fil du temps après avoir causé des dégâts lourds en terme de sur morbidité et surmortalité par cancer (plus de 20 000 cas supplémentaires de cancers du sein attendus chez les femmes britanniques de 50 à 64 ans à cause de l'usage des THS durant la dernière décennie) et de maladies cardiovasculaires.
Cette affaire illustre le caractère inapproprié dans ce cas du modèle de la pharmacovigilance fondé sur la notification spontanée. Seules des études épidémiologiques et des vastes essais randomisés post-AMM conduits par de très grandes équipes scientifiques ont été capables de faire la lumière sur ce problème. Ces équipes ont été co-financées par le National Heart Lung and Blood Institute des NIH et des laboratoires pharmaceutiques aux USA, par le Cancer Research UK et le MRC au Royaume Uni, par l'INSERM, la FRM et des laboratoires pharmaceutiques en France, et non par des équipes issues d'agences de sécurité sanitaire ou des systèmes de veille sanitaire. Ni la FDA, ni l'EMEA, ni l'AFSSAPS ne peuvent s'enorgueillir d'avoir participé au financement de ces récentes études.
Dans une certaine mesure, on peut reconnaître que ces résultats obtenus vis-à-vis des THS justifient la pratique récemment introduite en France de proposer des études post-AMM pour encadrer le lancement de nouveaux produits, tout en soulignant l'importance de les confier à des consortiums à haute valeur ajoutée sur le plan scientifique ».
4.2.3. Le retrait du Vioxx
Le développement après leur mise sur le marché à la fin des années 1990 des médicaments de la classe des anti-inflammatoires non-stéroïdiens (AINS) 36 ( * ) de la catégorie des « coxibs » (inhibiteurs de la cyclo-oxygénase (cox) de type 2 a été très rapide. Leur nouveauté, les avantages qu'on leur prêtait (moins d'effets indésirables) ainsi que d'intenses campagnes de promotion par les fabricants, sans parler de « prix d'appel » en milieu hospitalier, expliquent cet essor considérable.
En France, à la date du 1 er juillet 2004, deux coxibs utilisés par voie orale étaient commercialisés : le celecoxib (nom de spécialité Celebrex) et le rofécoxib (Vioxx), indiqués dans le traitement symptomatique de l'arthrose et de la polyarthrite rhumatoïde. Un troisième, le valdécoxib (Bextra) commercialisé en Amérique du Nord ne l'est pas en France.
Le laboratoire Merck-Sharp fabricant du Vioxx, de sa propre initiative en a suspendu et en fait cesser la commercialisation le 30 septembre 2004. L'analyse du problème peut être précisée à l'aide de la note de « mise au point sur la sécurité d'emploi des coxibs » diffusée par l'AFSSAPS le 1 er juillet 2004.
* Des questionnements et des doutes.
L'AFSSAPS rappelle tout d'abord les conditions du développement rapide des coxibs :
« Très rapidement après commercialisation, les coxibs auxquels on attribuait l'avantage de provoquer moins d'effets indésirables digestifs, ont atteint des niveaux de prescription élevés avec :
. un taux de prescription faites dans des indications non validées par l'autorisation de mise sur le marché (AMM)évalué à 11 % du total
. un taux de co-prescription avec les inhibiteurs de la pompe à protons (IPP) aussi important, voire plus, qu'avec les AINS dits « conventionnels », ceci alors qu'il n'existe aucune donnée clinique comparant l'association coxib + IPP à l'association AINS conventionnel + IPP
. de nombreuses notifications d'effets indésirables, parfois graves, amenant le système national de pharmacovigilance à s'interroger sur le profil de sécurité réel des coxibs ».
Elle aborde ensuite « ce qui a motivé le réexamen des données de sécurité des coxibs ».
« Parallèlement à la commercialisation des premiers coxibs, des publications ont fait penser que la réduction du risque de lésions gastro-intestinales serait moins importante que ce que laissaient supposer les études ayant conduit à l'AMM. Il est également apparu, dans des essais cliniques réalisés avec certains coxibs, que leur utilisation pouvait être associée à une augmentation du risque cardiovasculaire (hypertension artérielle, infarctus du myocarde, accidents vasculaires cérébraux).
Risque digestif : en France, les résultats du suivi national de pharmacovigilance des coxibs ont montré que le risque de lésions gastro-intestinales restait une préoccupation, en raison de la survenue d'accidents graves à type d'ulcères, de perforations et d'hémorragies digestives. De novembre 2000 à juin 2002, 320 cas ont été notifiés sous célécoxib (taux de notification : 3,5/10 000 patients-années) et, de juillet 2001 à juin 2002, 70 cas sous rofécoxib (taux de notification : 2,5/10 000 patients-années). Ces complications digestives, d'évolution parfois fatale, sont le plus souvent observées chez des patients présentant les facteurs de risque suivants :
. âge supérieur à 71 ans
. antécédents digestifs, en particulier ulcère
. prise concomitante d'aspirine, d'un autre anti-agrégant plaquettaire ou d'un anti-coagulant
Risque cardiovasculaire : au cours de ces mêmes périodes, des décès d'origine cardiovasculaire (infarctus du myocarde, arrêt cardiaque, trouble du rythme, insuffisance cardiaque, oedème aigu du poumon ou accident vasculaire cérébral) ont été notifiés au système national de pharmacovigilance : les taux de notification de ces décès sont respectivement de 2,6/100 000 patients-années, sous célécoxib et de 2,85/100 000 patients-années sous rofécoxib.
Risque cutané : suite à la survenue d'effets indésirables graves, sous valdécoxib et parécoxib, la question du risque cutané a été soulevée pour l'ensemble des coxibs.
Enfin, d'autres effets indésirables connus avec les AINS conventionnels, notamment des atteintes rénales, ont également été rapportés, confirmant que les coxibs présentent un profil de risque qualitativement identique à celui des AINS classiques.
L'AFSSAPS précise d'emblée (toujours le 1 er juillet 2004) que :
« Face à cette situation, la France a initié un arbitrage au niveau européen. Sur la base de l'ensemble des données disponibles, une réévaluation du rapport bénéfice/risque des coxibs a donc été entreprise en 2002, dont les résultats ont été avalisés par la Commission européenne, en avril 2004, et viennent d'être mis en ligne sur son site ».
* Elle présente alors « les résultats de la réévaluation européenne du rapport bénéfice/risque :
« Risque digestif
. l'utilisation des coxibs expose, de manière dose-dépendante, aux mêmes types d'effets indésirables gastro-intestinaux, parfois graves (ulcères, perforations, hémorragies) que les AINS conventionnels.
. d'un point de vue quantitatif, l'avantage des coxibs sur les AINS conventionnels n'apparaît pas constant, d'après les données disponibles. Pour le célécoxib, ces données sont en faveur d'un avantage par rapport au naproxène, mais la sécurité gastro-intestinale concernant les ulcères compliqués est similaire à celle de l'ibuprofène et du diclofénac. Quant au rofécoxib, il présente un avantage gastro-intestinal par rapport au naproxène et, dans une moindre mesure, par rapport au diclofénac et à l'ibuprofène. Il est toutefois difficile d'extrapoler les résultats obtenus avec ces comparateurs à l'ensemble des AINS conventionnels ; d'une part, parce que le risque relatif d'effets indésirables digestifs varie considérablement entre deux AINS conventionnés (dans un rapport qui peut aller de un à dix) et, d'autre part, parce qu'il dépend aussi des doses utilisées.
. Comme pour les autres AINS, le risque de complications digestives avec les coxibs est plus élevé chez les patients présentant un ou plusieurs des facteurs de risque suivants : âge avancé, prise concomitante d'un autre AINS ou d'aspirine (même à faibles doses), antécédents de lésions gastro-intestinales.
Risque cardiovasculaire
. Certaines données pré-cliniques suggèrent la possibilité d'une augmentation du risque cardiovasculaire, en particulier de survenue d'infarctus du myocarde ; cependant, en clinique, des résultats contradictoires ont souvent été observés. Au total, on ne peut pas exclure que les coxibs augmentent le risque cardiovasculaire par rapport aux AINS conventionnels.
. Comme les AINS conventionnels, les coxibs peuvent augmenter la pression sanguine artérielle chez certains patients (notamment en cas d'altération de la fonction rénale ou de prise de médicaments antihypertenseurs).
Risque cutané
. les coxibs, comme les AINS conventionnels, peuvent être la cause de réactions cutanées très rares, mais graves, à type de syndromes de Lyelle ou de Stevens-Johnson.
. En ce qui concerne le célécoxib, les études cliniques pré et post AMM montrent que le risque de réactions à type d'éruptions cutanées pourrait être plus élevé qu'avec les autres AINS.
Au total, il est nécessaire de renforcer les mises en garde et précautions d'emploi pour améliorer la sécurité des patients. Dans les indications autorisées, la balance bénéfice/risque des coxibs reste favorable à la condition de respecter les contre-indications et les mises en garde, en particulier celles qui viennent d'être ajoutées aux résumés des caractéristiques des produits concernés.
Ces nouvelles mesures visent la sécurité digestive, cardiovasculaire et cutanée ; cependant les coxibs présentent également d'autres types d'effets indésirables qui doivent être pris en compte même s'ils n'ont pas fait l'objet d'une réévaluation spécifique. Il convient notamment de rappeler le risque de survenue d'une insuffisance rénale aiguë comme avec d'autres médicaments connus pour inhiber la synthèse des prostaglandines ».
Cette réévaluation européenne s'est faite à la demande de la France dans des conditions qui, on le verra, ne semblent pas répondre à celles qui avaient été souhaitées au départ (méta-analyse). L'AFSSAPS concluait ainsi sa « mise au point » du 1 er juillet 2004 :
« Sur la base de l'ensemble des données disponibles à ce jour, la sécurité d'emploi des coxibs n'est pas remise en cause dans les indications qui ont été approuvées par l'AMM, tout en sachant qu'ils exposent, qualitativement, aux mêmes risques d'effets indésirables que les AINS conventionnels. En conséquence, les recommandations de bon usage des AINS s'appliquent également aux coxibs ».
* La mise au point de l'AFSSAPS avait été précédée et s'appuyait donc aussi sur l'avis de réévaluation de la commission de la transparence (du 16 juin 2004) rendu à la suite de la saisine conjointe de la direction de la sécurité sociale et de la D.G.S. du ministère en 2002 et suite à la réévaluation des coxibs par le comité d'experts de l'Agence européenne (EMEA) de février 2004.
Les conclusions de cet avis de la commission de la transparence annexées à la mise au point de l'AFSSAPS sont les suivantes (texte intégral) :
« Conclusions de la commission de la transparence
1) Service médical rendu :
La polyarthrite rhumatoïde et l'arthrose sont des maladies chroniques invalidantes.
Le rapport efficacité/effets indésirables est :
. moyen dans l'arthrose
. important dans la polyarthrite rhumatoïde
Il s'agit d'un traitement symptomatique.
Il existe de nombreuses alternatives thérapeutiques : l'ensemble des AINS (excepté les pyrazolés).
Le niveau de service médical rendu est important.
2) Amélioration du service médical rendu :
L'analyse des résultats disponibles montre que la meilleure tolérance digestive du Vioxx par rapport aux AINS anti-cox1 est minime en l'état actuel des données et du niveau de preuve présentés.
Il n'a pas été retenu de différence cardio-vasculaire notable par rapport aux AINS. Par conséquent, compte tenu d'une probable meilleure tolérance digestive, cette spécialité apporte une amélioration du service médical rendu mineure (niveau IV) par rapport aux AINS conventionnels.
3) Place dans la stratégie thérapeutique :
Arthrose : dans le cadre d'un traitement symptomatique de l'arthrose, il convient de débuter le traitement symptomatique par le paracétamol. En cas d'échec, les AINS sont prescrits, en commençant par des doses faibles. Ils doivent être réservés aux poussées douloureuses et ne pas être prescrits au long cours. La place du rofécoxib, comme celle de tous les AINS, se situe donc en deuxième intention. La prescription de cette spécialité aux patients à risque digestif (antécédents ulcéreux, sujet âgé, traitement anti-coagulant ...) justifie de prendre les mêmes précautions que celles recommandées pour l'ensemble des AINS.
Polyarthrite rhumatoïde : le traitement de la polyarthrite fait le plus souvent appel à des séquences successives de médicaments. Les anti-inflammatoires sont un traitement de première intention, associés aux traitements de fond. La prescription de cette spécialité aux patients à risque digestif (antécédents ulcéreux, sujet âgé, traitement anti-coagulant ...) justifie de prendre les mêmes précautions que celles recommandées pour l'ensemble des AINS.
4) Population cible :
Population cible dans l'arthrose : l'arthrose touche essentiellement les personnes de plus de 60 ans, les femmes plus fréquemment que les hommes (2 femmes pour1 homme). Sa prévalence en France est très mal connue et repose souvent sur des données anciennes et approximatives. Selon les données de l'enquête SPS du CREDES (rapport du Haut Comité de la Santé Publique, 2002) et les données du panel THALES (2001-2002), la population des arthrosiques symptomatiques serait de l'ordre de 4 à 5 millions de patients. Les AINS ne sont ni un traitement de première intention, ni un traitement au long cours de l'arthrose. Les seules données épidémiologiques disponibles sur le recours aux AINS sont celles du panel THALES 2001-2002. D'après ce panel, environ 60 à 65 % des arthrosiques seraient traités par AINS.
Population cible dans la polyarthrite rhumatoïde : sur la base des données épidémiologiques disponibles, la prévalence de la polyarthrite rhumatoïde peut être estimée à 130 000 à 240 000 patients. Selon les experts, la très grande majorité de ces patients est susceptible d'être traitée par des AINS.
5) Données d'utilisation (en annexe) :
6) Recommandations de la Commission de la transparence :
Avis favorable au maintien de l'inscription sur la liste des spécialités remboursables aux assurés sociaux et sur la liste des médicaments agréés à l'usage des collectivités et divers services publics.
Conditionnement : il est adapté aux conditions de prescription.
Taux de remboursement : 65 % »
* Le retrait lui-même
Le 30 septembre 2004, les autorités sanitaires sont informées simultanément, au niveau mondial, de la décision de retrait du fabricant. L'AFSSAPS publie le communiqué suivant le jour même :
Retrait mondial de la spécialité Vioxx
« L'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) vient d'être informée ce jour, comme toutes les autorités réglementaires concernées, de la décision des laboratoires Merck Sharp & Dohme-Chibret de l'arrêt mondial de la commercialisation de leur spécialité Vioxx (rofécoxib).
Cette décision intervient à la suite d'une analyse des résultats intermédiaires d'un essai clinique ayant mis en évidence un doublement du risque relatif d'événements cardiovasculaires (infarctus du myocarde et accidents vasculaires cérébraux) par rapport au placebo. L'agence note que cette étude :
- utilise la dose de 25 mg/jour, posologie maximale autorisée en France,
- est réalisée dans une indication en développement (polypose colique),
- que l'augmentation du risque n'est significative qu'au-delà de 18 mois de traitement continu, ce qui n'est le cas que d'un petit nombre de patients en France.
L'Agence rappelle que le rofécoxib est un anti-inflammatoire non-stéroïdien (AINS) de la famille des coxibs (inhibiteur sélectif de la cyclo-oxygénase-2) utilisé essentiellement dans le traitement symptomatique d'affections rhumatismales (arthrose et polyarthrite rhumatoïde).
Très rapidement après leur commercialisation, les coxibs, dont le rofécoxib, en raison d'un taux de prescription et de nombreuses notifications d'effets indésirables, ont fait l'objet d'un arbitrage au niveau européen, initié par la France en 2002. La réévaluation a confirmé le rapport bénéfice/risque favorable de cette classe an avril 2004 tout en renforçant les mises en garde et les précautions d'emploi pour limiter les effets indésirables gastro-intestinaux et cardio-vasculaires, notamment à long terme. En effet, l'hypothèse d'une élévation du risque cardiovasculaire avec les coxibs et plus particulièrement avec le rofécoxib a été évoquée à plusieurs reprises et la prudence recommandée chez les patients ayant des antécédents coronariens. Pour la première fois, une étude apporte des éléments complémentaires sur le sur-risque à long terme.
Aujourd'hui, le titulaire de l'autorisation de mise sur le marché (AMM), le laboratoire Merck Sharp & Dohme-Chibret, en accord avec l'Agence, procède au retrait des lots commercialisés. De même, les essais cliniques en cours sont suspendus.
Aussi, l'Agence recommande à tous les patients traités par Vioxx de consulter leur médecin pour modifier leur prise en charge thérapeutique. Cette démarche ne nécessite pas d'être réalisée dans l'urgence, compte tenu du fait que le risque de complication cardiovasculaire reste cependant faible et n'apparaît qu'à long terme ».
A ce communiqué, s'ajoute une mise au point détaillée sur la sécurité d'emploi des coxibs pour les praticiens et les patients. Les essais cliniques cités étaient entrepris dans le cadre de la recherche d'une nouvelle indication pour ce médicament et ne sont pas liés aux travaux que, par ailleurs, menait la FDA américaine (cf. infra sur les controverses mettant en cause la FDA).
Tout récemment (le 18 novembre 2004), le président directeur général de Merck a déclaré devant une instance du Sénat américain où il était auditionné, que Merck avait pris sa décision de retrait « quatre jours après avoir eu connaissance de la dernière étude montrant qu'une utilisation prolongée du produit faisait encourir au patient un risque cardiaque accru » (cité par Le Monde du 20 novembre 2004).
* Les études antérieures et la méta-analyse suisse
-- Plusieurs études portant sur les coxibs, et particulièrement sur le rofécoxib, avaient soulevé de sérieux doutes tant d'abord sur les avantages réels de meilleure tolérance par rapport aux AINS traditionnels, puis sur les risques cardiaques et circulatoires que pouvaient recéler les médicaments issus de ces nouvelles molécules dans une prise au long cours.
La revue Prescrire par la plume de son rédacteur en chef M. Gilles Bardelay, avait pris date d'une manière pour le moins prémonitoire dans un article de presse intitulé « Pharmacovigilance : d'une affaire à l'autre » (opinion dans le Monde daté du 11 septembre 2001). Traitant de l'affaire de la cérivastatine qui venait de trouver son épilogue (retrait du Staltor et du Cholstat le 8 août 2001) il détaillait ses doutes sur les « nouveaux AINS » dans des termes qui méritent d'être rappelés :
« Quelle sera la prochaine affaire ? Va-t-elle concerner un anti-inflammatoire non stéroïdien ?
Les anti-inflammatoires non stéroïdiens (AINS) sont de plus en plus souvent prescrits, dispensés et consommés, essentiellement comme antidouleur. Les campagnes de promotion des « nouveaux » AINS concentrent l'attention sur une hypothétique meilleure tolérance digestive. Mais qui parle actuellement des effets indésirables extradigestifs des AINS, auxquels n'échappent pas les substances les plus récentes ? Ces effets indésirables sont pourtant enregistrés aussi bien en cardiologie (insuffisances cardiaques et hypertensions artérielles iatrogènes) qu'en néphrologie (insuffisances rénales), en urologie (cystites, en particulier avec l'acide tiaprofénique), en dermatologie (réactions cutanées parfois graves au kétoprofène en gel ou crème), en gynécologie-obstétrique (infertilités, atteintes du nouveau-né à la suite d'une utilisation malgré la contre-indication au troisième trimestre de la grossesse), etc ... Sans parler de l'interaction avec les anticoagulants oraux.
Pour limiter les risques pris par les patients, les médecins et les pharmaciens mal documentés ne doivent pas attendre la prochaine affaire de pharmacovigilance : il leur faut réagir et se documenter correctement.
Une formation régulière à partir d'outils fiables, et donc forcément indépendants financièrement des firmes pharmaceutiques, est la condition indispensable pour que les médicaments soient utilisés au mieux : quand il faut et seulement quand il faut ; à la dose nécessaire et suffisante ; en pesant bien les risques possibles et les effets bénéfiques espérés ».
Le même auteur, qui avait ainsi pris date trois ans avant que le retrait brutal du Vioxx intervienne, rappelait sa position précédente dans un article de presse (Le Monde daté du 8 octobre 2004) où il mettait brutalement en cause la plupart des intervenants dans le domaine de l'information et de la formation à la thérapeutique, en première ligne les laboratoires pharmaceutiques, puis les professeurs de médecine, les autorités chargées de la fixation des prix ainsi que « la plupart des médias grand public ».
-- La publication le 5 novembre 2004 dans la revue médicale « The Lancet » de la méta-analyse d'une équipe de chercheurs de l'Université de Berne, en Suisse, a apporté un élément important, voire décisif, dans l'approche du problème posé par le Vioxx et surtout sur celui de son maintien sur le marché. L'équipe de l'Université de Berne, dirigé par M. P. Junï et le professeur Matthias Egger (département de médecine sociale et préventive et département de rhumatologie et d'immunologie clinique) a en effet procédé à une méta-analyse qui a consisté à passer en revue l'ensemble des données disponibles parues avant la décision de retrait du 30 septembre 2004 et accessibles sur les bases de données et dossiers informatisés de la FDA, soit 18 études sous contrôle placebo et 11 études d'observation.
Le texte de la dépêche d'Associated Press (en français) sur l'article du « Lancet » en donne le résumé suivant :
« Selon Peter Jüni, « l'analyse confirme ce que l'on suspectait, à savoir que l'on disposait dès la fin de 2000 de données prouvant un risque accru de maladies cardiovasculaires sous Vioxx ». A cette époque, 52 cas d'infarctus du myocarde se sont produits chez les 20.742 patients observés, dont 41 étaient traités avec Vioxx.
Les chercheurs suisses critiquent en particulier l'interprétation de l'étude « Vigor » de 2000, qui comparaît l'efficacité et les effets indésirables du rofecoxib (Vioxx) aec un autre antalgique, le naproxène, entre autres. L'étude avait mis en évidence des différences majeures dans les effets cardiovasculaires des médicaments. Les différences avaient alors été attribuées à des propriétés protectrices supposées du naproxène et non pas à des effets négatifs du Vioxx.
Les chercheurs bernois n'ont pour leur part trouvé aucune raison objective à cette interprétation. Dès lors, selon Matthias Egger, on aurait dû soupçonner une fréquence accrue des infarctus sous rofecoxib.
Selon l'équipe suisse, le groupe pharmaceutique Merck aurait dû ou pu retirer son médicament « plusieurs années » avant septembre 2004, c'est-à-dire dès qu'il a disposé des données utilisées par la méta-analyse. Or, à cette époque, Merck a confirmé la sécurité cardiovasculaire du Vioxx.
La méta-analyse a remis en cause d'autres déclarations du fabricant, notamment celle selon laquelle seuls les patients prenant du Vioxx depuis 18 mois au moins étaient concernés. « Les données montrent une augmentation de risque d'infarctus après seulement quelques mois de traitement et ce risque est indépendant de la dose », affirment les chercheurs bernois.
En outre, la méta-analyse met en évidence le fait que les études indépendantes du fabricant montrent de manière plus nette les effets négatifs du Vioxx. A l'avenir, selon les chercheurs, il s'impose que les données des études soient dépouillées de manière extérieure.
Les autorités de contrôle devraient revoir leurs procédures d'admission des médicaments. Les nouvelles données et informations relatives à un médicament devraient être reprises systématiquement dans la documentation et analysés en permanence. « Comme le montre l'exemple du Vioxx, c'est loin d'être le cas aujourd'hui », selon Junï. Seul un suivi continu des médicaments peut protéger la population.
« Dans le cas du Vioxx, un organisme indépendant devrait examiner les raisons pour lesquelles le fabricant et les autorités de contrôle n'ont pas assuré ce suivi dans la saisie des nouvelles données et informations », souligne encore le scientifique »..
* La position de la FDA
Le 3 novembre 2004, la FDA a rendu publique une étude selon laquelle le Vioxx pourrait, aux Etats-Unis, avoir été à l'origine de 27.785 infarctus du myocarde ou de décès par crise cardiaque entre 1999 et 2003.
La dépêche AFP du même jour (3 novembre à 18 h 30) en donne la le résumé ci-après :
« Quelque 53 % de ces accidents pourraient avoir été provoqués par la prise de doses standard de Vioxx, autant de cas qui auraient pu être évités si les patients avaient utilisé du Célébrex, l'anti-inflammatoire de Pfizer, souligne l'étude comparative du Dr David J. Graham, disponible sur le site internet de l'agence américaine chargée du contrôle des produits pharmaceutiques.
La prise d'une dose standard de Vioxx augmenterait les risques de 1,5 fois et la prise de fortes doses de 3,7 fois, précise l'étude menée entre 1999 et 2001 sur des patients californiens âgés de 18 à 84 ans.
Entre 1999 et 2003, environ 92,7 millions de prescriptions de Vioxx ont été faites aux Etats-Unis, dont 17,6 % de fortes doses, c'est-à-dire supérieures à 25 milligrammes par jour.
Le nombre d'infarctus et de décès attribuables à la prise de doses standard de Vioxx a été sur cette période de 14.845 et celui lié à des doses fortes de 12.940.
C'est pourquoi « l'utilisation de fortes doses de rofecoxib (Vioxx) devrait être abandonnée et les faibles doses ne devraient pas être utilisées par les médecins ou leurs patients », préconise le Dr Graham.
« Si les faibles doses devaient rester sur le marché, médecins et patients devraient comprendre que le risque d'infarctus du myocarde et de décès par crise cardiaque est augmenté substantiellement et qu'il existe des alternatives moins risquées », ajoute-t-il.
L'agence fédérale attaque également les affirmations du fabricant du Vioxx, Merck, selon lesquelles les risques supérieurs liés à la prise du médicament mis en évidence par une étude le comparant à un autre anti-inflammatoire, le Naproxen, résultaient d'un effet protecteur du Naproxen encore jamais reconnu ».
Or, souligne la FDA dans son étude, « aucun effet protecteur (du Naproxen contre les risques cardiaques) n'a été démontré ».
Dans son édition de lundi, le Wall Street Journal affirmait que Merck était au courant des dangers encourus par les patients utilisant le Vioxx et aurait lutté pendant des années pour étouffer les inquiétudes sur les dangers du médicament ».
Ces révélations successives interpellent évidemment très durement le fabricant (Merck) dont la vigilance n'a pas été à la mesure du risque qu'il pouvait soupçonner depuis longtemps et ce par plusieurs études différentes. Le fait que le Vioxx, lancé et promu commercialement avec des moyens importants, était à l'origine de 11 % du chiffre d'affaires du groupe en 2003 (1,9 milliards d'euros) constitue un élément dont il a pu être excessivement tenu compte. Il convient d'ailleurs de rappeler qu'au moment de la prise de décision du retrait du Vioxx, le souci s'est clairement manifesté de prévenir en priorité les actionnaires du groupe Merck et les autorités boursières, l'impératif de santé publique paraissant passer au second plan.
Mais la FDA elle-même se retrouve en position délicate pour n'avoir pas elle-même réexaminé le cas d'un médicament dont les risques apparaissaient à travers ces études, au-delà de l'AMM qu'elle avait donnée. En outre, une polémique très violente se développe depuis plusieurs semaines à partir de suspicions de pressions, au sein même de la FDA sur le directeur adjoint du service chargé d'évaluer l'innocuité des médicaments (le Dr Graham). Le président de la commission des finances du Sénat américain procède depuis le début du mois de novembre 2004 à des auditions sur ce sujet. De son côté, le Wall Street Journal consacre, en suivant cette actualité de très près, de nombreux articles à cette affaire. Tout en se gardant de tirer des conclusions hâtives sur ces aspects de l'affaire, la mise en cause de la FDA, de son fonctionnement et de la philosophie de son action incitent à se poser des questions de principe sur les mécanismes d'autorisation et de suivi (pharmacovigilance). Un enseignement doit être tiré de tels événements. Avant d'y procéder, plusieurs précisions restent à apporter sur les « aspects collatéraux » de la décision de retrait du Vioxx, car l'ampleur du problème a immédiatement soulevé des questions de parallélisme avec d'autres médicaments de cette classe thérapeutique à commencer par le besoin de remplacement.
* On a noté les prescriptions de l'AFSSAPS lors du retrait (cf. supra, orientations vers le paracétamol, médicament « classique » et ancien). La réaction naturelle a été par ailleurs une mise en cause sur d'autres « coxibs », mais aussi paradoxalement une tentation de se reporter sur ceux qui restaient en vente. C'est évidemment le cas du Celebrex (le seul en France, le Bextra quant à lui étant autorisé aux Etats-Unis, mais pas en France. Le jugement du Pr Nicholas Moore 37 ( * ) (Service de pharmaco-épidémiologie - Bordeaux II), chargé de la surveillance des coxibs en France synthétise le problème : « Si le risque cardio-vasculaire, comme on le croit, est lié à la sélectivité des coxibs, le Bextra, comme le Vioxx, est justement très sélectif et à l'inverse, le Celebrex, qui n'est pas plus sélectif que le Voltarène, est indemne d'effets secondaires sur le coeur ».
L'auteur de l'article, J.M. Bader, fait suivre cette citation de la remarque suivante : « Les coxibs, comme le martèle depuis 2001 la revue médicale Prescrire, n'auraient donc aucun avantage par rapport aux anciens médicaments associés à un protecteur gastrique et seraient en tout cas beaucoup plus chers. Six firmes dont Novartis, Yamanouchi et Glaxosmithkline, ont encore des molécules de la famille des coxibs à mettre sur le marché ».
En ce qui concerne la France, pendant le mois qui a suivi le retrait du Vioxx, c'est-à-dire le mois d'octobre 2004, les ventes de Celebrex ont augmenté de 30 %, ce qui compte tenu du besoin et de la tentation de report n'est qu'apparemment paradoxal.
La version révisée des données préliminaires communiquées en novembre 2004 par le Dr Graham (FDA) a été publiée par la revue britannique « The Lancet » le 25 janvier 2005 et fait état d'un total de 88.000 à 140.000 cas supplémentaires de « maladie cardiaque sévère » aux Etats-Unis entre 1999 et 2004 (période de commercialisation). Ces chiffres, résultats d'une extrapolation réalisée à partir de deux essais cliniques restent entachés d'une « grande marge d'incertitude » selon les commentateurs spécialisés 38 ( * ) . Le Pr Nicholas Moore (déjà cité) qui doit publier prochainement l'étude attendue sur la France, considère de son côté « que cette étude est fragile et manque de puissance » 39 ( * ) .
4.3. Quels enseignements pour les agences du médicament ?
L'extrême acuité d'une question dont le développement ne fait sans doute que commencer avec le Vioxx ne permet pas de tirer des « enseignements stabilisés » et en tout cas pérennisés. Mais sa remise en perspective avec les problèmes de fond qui ont été identifiés autorise à pointer les difficultés et, d'autre part, à rappeler les impératifs auxquels les procédures et les instances doivent rester soumises.
4.3.1. Des difficultés croissantes
* Identifiées depuis longtemps pour la plupart, elles sont devenues plus aigues au fur et à mesure des développements scientifiques et technologiques, notamment de la puissance des médicaments, de la capacité à en mesurer les effets, de la lourdeur et de la longueur des recherches, de la complexité des procédures d'expertises, ainsi que celles d'autorisation de mise sur le marché. En outre, l'évaluation, les exigences de sécurité se situent à un niveau de plus en plus élevé alors qu'on attend en même temps une efficacité de plus en plus grande.
Or, on se trouve ici dans un domaine spécifique avec la nécessité de tirer des éléments de comparaison exprimés en terme de souffrance, maladie et mort. Le rapport n'est pas en termes de « coût/avantage », mais cela doit être souligné de « bénéfice/risque ». Si le risque zéro n'existe nulle part, dans le domaine pharmaceutique le risque non négligeable est généralement avéré, en rapport avec le bénéfice médical attendu. Il n'est donc pas anormal que des médicaments puissent être risqués, voire dangereux (certains anticancéreux par exemple). Le point d'équilibre est donc difficile à déterminer et l'évolution naturelle renforce cette difficulté.
* Le poids de la dimension financière des exigences du marché n'est pas moindre et, on le voit avec le cas du Vioxx, la stratégie de recherche et de développement commercial d'un nouveau médicament a éclipsé brutalement le critère premier qui devait rester celui du bénéfice médical ajouté.
On retire l'impression fort inquiétante que la procédure du retrait du Vioxx par Merck devait surtout satisfaire les procédures boursières en réservant, comme la SEC le prévoit, la primeur de l'information aux actionnaires.
* Enfin, ne doivent pas être oubliées non plus les contraintes que représentent pour l'industrie pharmaceutique la longueur des procédures d'AMM, le coût des recherches et des besoins cliniques qui justement doivent apporter davantage de garanties que par le passé, la durée de vie commerciale limitée des médicaments et la judiciarisation peut décourager l'innovation scientifique. On aura garde d'oublier aussi le poids de la promotion auprès du corps médial qui s'illustre en France entre autres par l'existence d'un réseau de 23.000 visiteurs médicaux pour 206.400 médecins (de ville et hospitaliers).
4.3.2. Les impératifs confirmés
4.3.2.1. Les principes généraux
* Les principes fondamentaux des procédures d'expertise d'autorisation et de surveillance des médicaments ont naturellement des objectifs de sécurité et d'efficacité qui sont par essence universels, mais leur délivrance selon les cultures et les époques peut enregistrer des variations. En plein développement de la crise du Vioxx, l'adjointe au directeur général de l'AFSSAPS, Mme Emmanuelle Wargon, indiquait ainsi (dépêche AFP du 24 novembre 2004) : «L'AFSSAPS est au service de la sécurité sanitaire et de la protection des patients. La FDA dit explicitement qu'elle est au service de l'innovation, de la rapidité d'accès des produits au marché, ce qui est un peu différent ».
* Dès lors, ce sont les conceptions mêmes de la fonction des autorités investies des procédures d'autorisation et de surveillance qui ne se recoupent pas exactement. Une telle distinction apparaît d'ailleurs au sein même de l'Union européenne lorsque l'on observe une tendance récente « à faire vite » et peut conduire à une concurrence entre les agences nationales dans le cadre de la procédure de reconnaissance mutuelle. On a ainsi toutes les raisons d'être inquiets pour l'avenir sachant que lorsqu'il y a vote, chaque Etat-membre dispose du même poids.
* S'agissant de la sécurité de la procédure d'AMM, la conception et les pratiques du dispositif français à travers l'AFSSAPS et dans le cadre européen, en partie inspirée précisément de l'expertise et des procédures françaises, doivent être confortées et non remises en cause. Il y a lieu de rappeler que les crises majeures enregistrés à ce jour n'ont pas impliqué directement les autorités françaises dont la prudence où le jugement ont été moins mis en cause que d'autres (cérivastatine, THS et Vioxx).
Des exigences de renforcement des essais cliniques peuvent être recevables, mais les limites physiques, financières et cliniques risquent d'être rapidement atteintes compte tenu du niveau déjà élevé. On sait que la majorité des essais sont plutôt faits sur des hommes majeurs, non âgés et indemnes d'affections autres que celles visées par le médicament. Le spectre est donc à élargir ; mais on devine sans peine les difficultés, surtout éthiques, que cela pose avant même toute considération méthodologique et financière. C'est précisément lorsque la diffusion d'un médicament dépasse nettement les quelques milliers d'essais pour être ensuite répandu dans le public, que l'on peut « espérer » réparer des effets indésirables ou des insuffisances qui ne sont pas identifiables avant.
4.3.2.2. La transparence des essais cliniques
La transparence des essais cliniques est une exigence essentielle qui avait été formulée de différents côtés, notamment aux Etats-Unis, et à différentes reprises. Pour la satisfaire enfin, il aura fallu le bouleversement entraîné par l'affaire du Vioxx. En effet, le 6 janvier 2005, la fédération internationale des associations de producteurs pharmaceutiques (IFPMA) avec les trois fédérations continentales de l'industrie pharmaceutique (Amérique - Europe - Japon) ont dans une démarche commune annoncé qu'elles « s'engagent à augmenter la transparence des essais cliniques menés par leurs membres ».
Ces associations reconnaissent l'importance de la diffusion des informations en la matière pour les praticiens des soins de santé et pour les patients. Les informations sur les essais cliniques en cours, qui doivent permettre de déterminer les effets thérapeutiques d'un médicament, devront être consignées dans un registre accessible au public, gratuitement, dans les 21 jours suivant le démarrage de ces essais. Ce registre contiendra les informations de base pour chaque essai, de sorte que les patients et leurs médecins disposent de toutes les données nécessaires sur la manière d'y participer. Les résultats des essais cliniques menés sur des médicaments qui ont obtenu une autorisation de mise sur le marché et qui sont commercialisés, dans au moins un pays, devront ensuite être consignés dans une base de données également accessible au public. Lorsque ces résultats seront publiés dans un journal médical, la base de données devra inclure une citation de cet article ou un lien vers cet article.
En France, le LEEM (Les entreprises du médicament) a confirmé et précisé le 11 janvier 2005 la mise en oeuvre de ces engagements pour ce qui le concerne en indiquant que la création du « registre d'essais cliniques » serait mis en place le 1 er juillet 2005.
4.3.2.3. La pharmacovigilance
C'est donc en outre par un développement d'une pharmacovigilance systématisée, d'un suivi post-AMM formalisé à rendez-vous fixes et contraignants que l'on peut espérer réaliser des progrès substantiels. On a vu également que la iatrogénie médicamenteuse « classique » exigeait elle aussi de nouveaux efforts (cf. supra). Cela implique que la formation et l'information du corps médical soient renforcées par rapport à celles observées aujourd'hui en France.
4.3.2.4. L'information du corps médical
Ce point étant naturellement important, mais éloigné de l'évaluation de la loi de 1998 qui est l'objet du présent rapport, on se gardera de tout diagnostic définitif. Il convient simplement de rappeler que l'on ne saurait accepter que l'essentiel de l'information thérapeutique soit distribuée par les 23.000 visiteurs médicaux que la France compte aujourd'hui. C'est pour le moins unilatéral et insuffisant.
L'enseignement de la thérapeutique n'a pas dans le milieu hospitalo-universitaire la place qu'il devrait avoir. Alors que la puissance des médicaments se développe à un rythme accéléré, il devient encore plus urgent que les responsables mettent un terme à cette lacune.
L'AFSSAPS elle-même a une perspective de développement considérable et son intervention est tout à fait nécessaire et attendue : les différents intervenants s'accordent à reconnaître que l'Agence dispose sinon de « trésors », du moins de ressources considérables qui sont actuellement inexploités et dont les médecins, généralistes notamment, pourraient tirer le plus grand profit. On peut espérer que la résolution des problèmes informatiques (cf. supra) permettra d'atteindre cet objectif.
4.3.2.5. La position des experts
La résolution des conflits d'intérêts s'apparente rapidement à la quadrature du cercle. On ne reviendra pas sur les rapports entre l'expertise interne et l'expertise externe qui a déjà été traitée (cf. supra) et qui n'apporte pas de réponse au problème ; la situation des pays dont les Etats-Unis, où l'expertise interne a une beaucoup plus grande place montre que les conflits d'intérêts y posent des difficultés considérables.
La publication des listes de déclarations d'intérêts des experts membres de comités spécialisés entraîne des remarques critiques, mais c'est précisément l'absence de déclarations antérieures qui est d'abord la plus critiquable ! Le respect de la déontologie élémentaire passe d'abord par la transparence.
La possibilité de recourir à un vivier fourni d'experts nombreux et compétents est certainement l'un des moyens d'éviter des dérives. La promotion de la fonction d'expertise au niveau des curriculum vitae des chercheurs dans ce domaine scientifique aussi, comme dans d'autres où la situation est grave (toxicologie), est un impératif ; l'expertise passe malheureusement souvent après la prise en compte de publications dont le statut et la valeur mériteraient dans certains cas d'être sérieusement vérifiés. La responsabilité en incombe ici directement aux autorités universitaires.
4.3.2.6. La dimension économique de la recherche pharmaceutique
La récente fusion absorption Aventis/Sanofi-Synthelabo a permis de noter que la première de ces entités, elle-même issue de fusions successives, n'indiquait pas un nombre de projets de nouveaux médicaments en rapport avec ce que ces regroupements antérieurs auraient pu laisser supposer. C'est la une illustration d'une manière générale des limites de l'argument que l'on avance systématiquement en faveur de la contribution de « grands groupes ».
La réduction du nombre de nouveaux médicaments lancés au cours de la dernière décennie est aussi à remarquer, dans le domaine des antibiotiques par exemple.
La diminution des prix, la baisse des prix de remboursement, les menaces de procès systématiques pèsent aussi sur les perspectives. Or une crise considérable comme celle du Vioxx est de nature à réduire encore les perspectives de développement donc de recherche. Certes des perspectives nouvelles comme celles des bio-médicaments peuvent apporter des éléments favorables à une évolution inverse, mais leur importance reste à apprécier.
Aussi, mais il s'agit là d'une observation plus que d'une prescription, on peut craindre un ralentissement de nouvelles découvertes pharmaceutiques, ce qui constitue une préoccupation sérieuse qui doit rentrer en ligne de compte dans l'orientation des politiques, tout en prenant place dans une vision sanitaire plus large intégrant des actions de prévention fortes et organisées.
V. LES RISQUES ÉMERGENTS
* Les dispositifs juridiques relatifs à l'expertise, la diffusion, la dispensation et la surveillance du médicament et des produits de santé se sont mis en place en France dans des conditions satisfaisantes. Leur perfectionnement se poursuit, même si certains aspects doivent faire l'objet d'efforts soutenus, la pharmacovigilance notamment. Cependant, dans le même temps, on observe des évolutions factuelles et juridiques, en particulier dans le cadre européen qui recèlent de véritables risques.
En France, le cadre juridique de l'activité de distribution du médicament et son contrôle ont permis depuis de longues années d'améliorer encore la sécurité et l'efficacité : présence et contrôle effectif par le pharmacien de la distribution des médicaments dans l'officine, moyen d'identification directe de la fonction des personnels au contact des clients, fonctionnement excellent des procédures de rappel de lots ... La valeur de ce réseau de distribution à partir du fabricant et à travers les grossistes-répartiteurs est indiscutable et constitue une référence mondiale. Elle est la contrepartie d'une organisation exigeante fondée sur un monopole lui-même ancré dans l'exigence de santé publique.
A une époque où on invoque souvent le « principe de précaution », où l'on s'inquiète à juste titre des effets indésirables et des interactions des médicaments, la valeur irremplaçable du dialogue avec le dispensateur autorisé du médicament dans l'officine se confirme quotidiennement. Il est donc pour le moins paradoxal que l'on se propose, par les biais juridiques ou matériels les plus spécieux et les montages les plus inextricables, de saborder un système qui fait ses preuves. La nécessité de ce dispositif et la justesse des principes simples et efficaces sur lesquels il est fondé se vérifie à chaque comparaison internationale.
* L'évaluation de la loi de 1998 perdrait toute signification si on basculait vers un système fondé sur internet. Le schéma est en rupture totale avec les principes actuels qui apportent de réelles garanties. Cette nouvelle organisation pourrait se résumer en quelques lignes : il s'agit de la dispensation du médicament à un « cyberclient » par un « cyberpharmacien » installé dans un pays lointain, éventuellement dans une langue étrangère sur la base d'une ordonnance dont tous les éléments essentiels (indication, posologie, dosage, conditionnement etc ...) peuvent varier d'un pays à l'autre. Bien sûr, l'échange entre le « cyberpharmacien » (en espérant que celui-ci n'est pas un simple vendeur sans qualification) et le « cyberpatient » (on se sera assuré que celui-ci n'est pas une tierce personne par substitution d'identité), devrait prendre en compte les prescriptions préexistantes et l'administration concomitante d'autres médicaments ; en cas de difficultés ou d'incidents, le « cyberpatient » devrait aussi recontacter le même « cyberpharmacien » pour lui retourner ou échanger un médicament ...
On mesure la multiplication des risques produits par de telles procédures sous couvert de modernité, de rapidité, d'efficacité et de rationalité financière.
Aux esprits « rétrogrades » que ce tableau inquiéterait, on peut répondre que les procédures seront « sécurisées », que la dialogue par Internet est banalisé et que la livraison par portage n'est pas plus un problème pour les médicaments que pour les pizzas, à moins que l'on exige, comme ce serait le cas dans certains pays, que celle-ci soit assurée par « du personnel de l'officine ». Les procédures sécurisées permettront aussi, bien entendu, de s'assurer que l'ordonnance et l'identité du patient sont exactes et non falsifiées (un copié-collé etc ...) et que le caractère personnel et confidentiel des données médicales échangées au cours du « cyber dialogue » sera sauvegardé, par le recours aux techniques de biométrie (vérification de l'identité et de la qualité des interlocuteurs) tout cela en conformité avec la loi informatique et liberté ...
Rejetons aussi pour argument non fondé le cas des personnes âgées et/ou malades ayant des difficultés à se déplacer, mais qui curieusement n'en n'auraient aucune pour entretenir un « dialogue cybernétique » précis et complet, dépourvu de risque d'erreur et d'ambiguïté, éventuellement dans une langue étrangère.
Cette perspective, qui peut rapidement tomber dans l'anticipation la plus noire n'a rien de fantaisiste ou même de lointaine. Elle s'inscrit dans la logique de dispositions récentes prises par l'Allemagne. Dans ce pays voisin, la recherche systématique et justifiée d'économies au niveau de l'assurance maladie semble avoir entraîné l'affranchissement des règles fondamentales en matière de dispensation du médicament. Encore plus inquiétant, certains risques identifiés depuis quelque temps déjà deviennent réalité grâce à la mondialisation et à la banalisation du médicament avec les contrefaçons et les faux médicaments. Le trafic de médicaments authentiques ou contrefaits peut s'ajouter à la liste des activités frauduleuses de la mondialisation « noire ».
5.1. Des perspectives incertaines au niveau européen
5.1.1. La fragilité du fonctionnement actuel
5.1.1.1. Les interrogations sur les mécanismes d'AMM
* Le fonctionnement relativement satisfaisant atteint actuellement par l'Agence européenne et le réseau des agences nationales n'est pas un gage de réussite pour l'avenir, même proche.
Tout d'abord, certains épisodes ont montré les faiblesses et surtout les lenteurs de réaction de l'EAMA de Londres. Ce qui était perceptible avec l'affaire de la cérivastatine (Cholstat et Staltor) l'est encore bien davantage avec celle du Vioxx. Saisie en 2002 par l'AFSSAPS, l'EAMA n'a pas procédé à la méta analyse que l'équipe de chercheurs de l'Université de Berne a réalisé avec des moyens dont on peut penser qu'ils ne sont pas supérieurs. L'EAMA a demandé aux principales agences nationales de participer à la réévaluation du Vioxx et celle-ci ne s'est pas révélée décisive. De même, sa réactivité à la suite du retrait intervenu le 30 septembre 2004 ne paraît pas à la hauteur des enjeux.
-- Les délais maxima imposés par les procédures d'AMM (210 jours pour la centralisée et la nationale) ont pu être maintenus à un niveau satisfaisant pour la qualité de l'expertise scientifique lors de l'élaboration de la Directive 2004/27/CE et au Règlement CE n° 726/2004, mais on a pu remarquer des pressions pour raccourcir ces délais du côté des fabricants comme de la Commission elle-même, qui proposait 150 jours pour la procédure nationale. Une exception a été consentie pour la procédure centralisée à 150 jours dans le cas « d'un médicament présentant un intérêt majeur du point de vue de la santé publique et notamment du point de vue de l'innovation thérapeutique ». Il en a été de même pour la réévaluation de l'AMM au terme d'un délai de cinq ans qui a été maintenu, mais dont la Commission proposait la suppression, au profit d'une AMM illimitée.
-- La procédure de l'AMM par reconnaissance mutuelle qui avait été conçue pour avoir un caractère transitoire en attendant la montée en puissance de la procédure d'AMM centralisée a été en fait pérennisée ; or, contrairement à celle-ci, elle est moins transparente.
-- Plus inquiétant est le fait que les fabricants ont le pouvoir de choisir en fait le « pays rapporteur » donc l'agence qui examinera sa demande. Ils sont naturellement tentés de choisir celle qui l'examinera le plus vite et qui manifestera un niveau d'exigence moins élevé. Si l'on garde à l'esprit que les ressources des agences nationales proviennent pour l'essentiel des redevances versées par les fabricants lors du départ de leurs demandes, la pression sur les agences pour un examen rapide et compréhensif n'en est que plus forte. Or, la disparité des conditions d'examen et des capacités d'expertise n'est pas niable. Ce qui pouvait être un problème limité à Quinze a toutes les chances de devenir préoccupant à Vingt-cinq, sachant qu'on ne saurait reprocher aux nouveaux Etats-membres de ne pas disposer de l'expertise et de l'expérience qui ne pouvait par définition être la leur avant 1989. Certes, dans le cadre de procédure de « pré adhésion », les agences nationales ont été amenées à développer une coopération systématique avec les instances homologues des pays alors candidats pour leur permettre d'atteindre un niveau opérationnel dès l'adhésion (Grande-Bretagne avec République Tchèque, France avec Pologne, etc ...). Il serait toutefois logique de s'assurer que les objectifs ont été atteints.
A ce sujet, il doit être rappelé que dans les instances de l'EAMA où l'on est amené à prendre des décisions collectives, les représentants de chaque Etat-membre disposent de la même voix, quelle que soit sa population et sa production pharmaceutique.
-- L'environnement organisationnel est également devenu « concurrentiel » pour le contrôle des vaccins. En effet, dans le cadre de la procédure européenne de libération des lots pour les produits immunologiques, le fabricant peut choisir le laboratoire de contrôle qu'il souhaite parmi tous les laboratoires officiels européens.
5.1.1.2. La dimension européenne des inspections
L'harmonisation effective des systèmes de sécurité sanitaire européens pour les médicaments, mais aussi pour les vaccins et les dispositifs médicaux, est une condition à la réalisation d'objectifs minimaux dans ce domaine pourtant essentiel. Le constat fait par l'Inspection générale des finances et l'Inspection générale des affaires sociales dans leur audit daté de décembre 2002 (cf. supra) est de nature à nourrir des inquiétudes (op.cit page 34) :
« Bien qu'elle n'ait pas documenté les garanties apportées par les autres Etats-membres, la mission constate toutefois que dans la réalité, aucun mécanisme n'est réellement prévu pour garantir l'application effective des principes généraux de reconnaissance d'équivalence des systèmes, notamment en matière d'inspection.
La mission a pu illustrer cette question à partir des exemples suivants.
• Dans le secteur des
médicaments
chimiques
, les directives successives prévoient depuis 1993 une
équivalence entre les inspections de chaque Etat-membre, seul
compétent sur son territoire. Ainsi, il ne peut être
envisagé, par exemple, que la France inspecter un site de fabrication
espagnol, même en cas de signalement sur un médicament
fabriqué en Espagne et circulant librement sur l'ensemble de l'espace
européen.
Or, l'équivalence des inspections telle que prévue dans les textes est pour partie théorique. En effet, lors de la préparation d'un accord de reconnaissance mutuelle avec un pays tiers à l'Union, des audits préalables de chacune des inspections des Etats de l'Union ont mis en lumière des disparités dans la qualité du fonctionnement des inspections et pour un des Etats de l'Union des insuffisances si importantes que l'accord se trouve bloqué.
• S'agissant des
produits pour lesquels
les directives ne prévoient pas une telle équivalence
, la
France peut être conduite à diligenter des inspections sur des
sites de fabrication dans les Etats de l'Union. Dans sa pratique, cette
démarche est difficile à mettre en oeuvre puisqu'elle suppose une
action diplomatique aux effets aléatoires. Face à un tel enjeu,
les modalités de coopération entre Etats devraient être
renforcées pour permettre une harmonisation effective des pratiques
entre Etats.
Par ailleurs, au-delà de l'espace européen, une réelle mutualisation des moyens doit s'opérer pour démultiplier les capacités d'inspection dans des pays tiers à l'Union. En ce sens, les réflexions en cours pour faire émerger des bases de données partagées des rapports produits par les inspections des pays de l'Union constituent certainement une voie à encourager.
Une limite du champ européen porte sur l'articulation entre la DEQM et l'AFSSAPS. En effet, l'Agence apporte son concours aux travaux de la DEQM, notamment pour l'organisation d'inspections de sites de fabrication de matières premières à usage pharmaceutique, sans pour autant que la base de données des CEP (certificats européens de conformité à la pharmacopée) détenue par la DEQM à Strasbourg soit accessible à l'Agence dans le cadre de ses travaux nationaux.
• Dans le domaine
des dispositifs
médicaux
, la même problématique liée à
l'effectivité de l'équivalence des organismes notifiés des
différents Etats membre se pose.
• En matière de
laboratoires de
contrôle
, la France jouant un rôle leader, la question de
l'équivalence des contrôles en laboratoire est sans doute moins
aiguë. De plus, un système d'audits mutuels des différents
laboratoires a été mis en place et des procédures de
certification par des autorités indépendantes sont en cours dans
tous les pays de l'Union.
• Enfin, la mission a pu relever
les
difficultés de mise en oeuvre de systèmes d'information communs
aux Etats
».
Le même audit conclut sur ce point :
« L'émergence d'un système européen avec les risques vus plus haut d'une sécurité sanitaire a minima a justifié jusqu'ici la forte implication de l'Agence à un niveau à la fois technique et stratégique afin d'obtenir des garanties de haut niveau appliquées également à l'ensemble des pays de l'Union ».
Enfin, ce même document (page 60) traitant le problème de la nécessité pour l'Agence elle-même de préciser l'implication de celle-ci dans les travaux au sein de l'Union européenne (cf. supra) confirme explicitement les motifs d'inquiétude qui ne se sont pas démentis depuis :
« De plus, comme la mission a pu le montrer, de réels sujets d'inquiétude peuvent d'ores et déjà être identifiés dans l'Union, compte tenu de la liberté de circulation des produits et de l'absence d'harmonisation effective des pratiques de sécurisation des produits de santé entre Etats. Des propositions de la France sur ces questions seraient de nature à accroître l'investissement européen de l'Agence ».
5.1.2. Des tentatives de banalisation du médicament
De différents horizons des tentatives se multiplient pour ôter au médicament son statut spécifique qui a été érigé pour des raisons évidentes. Les traités européens ont d'ailleurs ab initio reconnu cette spécificité et les restrictions naturelles qu'elle implique, dans la distribution notamment. Les dispositifs juridiques et administratifs qui en découlent sont généralement cohérents et précis, notamment en ce qui concerne la France. Cela ne décourage pas pour autant des milieux qui n'ont de cesse, par tous les biais imaginables, de faire abolir ces dispositifs.
* Le cas des importations parallèles : pratique légale qui s'est établie au sein de l'Union européenne et qui en France a fait récemment l'objet d'un texte réglementaire les organisant (janvier 2004), les importations parallèles (en amont de la distribution au public) par les grossistes répartiteurs ou les pharmaciens permettent de s'approvisionner dans des Etats-membres où les médicaments considérés sont nettement moins chers.
Il ne s'agit pas ici de revenir sur ce régime juridique et les raisons du développement de cette pratique légale. Le réel problème qui se pose dans ce domaine est celui du déconditionnement des médicaments et même de la « déblistérisation » des plaquettes de produits qu'autorisent les dispositions européennes avec tous les re-conditionnements possibles. Cela interdit en pratique la procédure de rappel des lots et ouvre de remarquables possibilités, à la contrefaçon notamment.
* Lors de l'élaboration de la Directive n° 2004/27/CE et du Règlement 726/2004, la Commission européenne, reprenant des demandes de firmes pharmaceutiques, avait proposé d'autoriser pour les médicaments soumis à prescription médicale une publicité déguisée en l'espèce sous forme « d'information du public ». La publicité pour ces médicaments étant interdite dans les pays de l'Union, cette proposition a rencontré une vive opposition au Parlement européen. De leur côté, la plupart des ministres des Etats-membres avait également exclu une telle possibilité. Cette tentative témoigne néanmoins de l'opiniâtreté à attaquer de front des règles évidentes de santé publique, l'argument étant généralement que « cela se pratique hors d'Europe ».
* Tout récemment, le groupe de travail présidé par M. Michel Camdessus, à la demande de M. Nicolas Sarkozy, alors Ministre de l'économie, des finances et de l'industrie, a remis un rapport intitulé « Le sursaut - vers une nouvelle croissance pour la France ». Dans un chapitre VI (Assurer l'efficacité des marchés des biens et des services), un développement dont le titre révélateur est « Supprimer les réglementations qui visent essentiellement à protéger les situations acquises » s'attaque à des secteurs qu'il s'agit ainsi de commencer par stigmatiser, parmi lesquels les taxis parisiens ... et les pharmacies. Il est rappelé en tête du paragraphe qu'il s'agit de « quelques réglementations à faire évoluer en prenant davantage en compte l'intérêt de tous les citoyens ». Le texte place donc sous un chapeau commun des situations fort diverses, totalement hétérogènes, en précisant : « On notera ici quelques exemples qui nous placent dans une situation particulièrement atypique par rapport à nos partenaires » . Le passage relatif à la pharmacie indique sans ambages la direction :
« Le cas des pharmacies est également spécifique à la France. Si partout en Europe l'activité de pharmacie est encadrée, cet encadrement est beaucoup plus strict en France puisqu'il concerne la répartition du capital : un pharmacien ne pouvant être propriétaire de plus d'une officine, la constitution d'une chaîne officinale permettant une diminution des prix est impossible. Dans le même secteur, la France est l'un des rares pays à étendre le monopole de vente des médicaments sur prescription aux médicaments hors prescription, ce qui empêche une distribution plus large accompagnée d'une diminution des prix ».
Rien ne devrait donc entraver les perspectives de pression sur les prix, qui sont d'ailleurs très relatives dans les situations d'oligopoles résultant de concentrations abusives. L'exercice personnel de la pharmacie devrait donc s'effacer face aux exigences de la « libéralisation » du marché. La réalité est fortement malmenée, notamment lorsqu'on indique qu'on est là dans une « situation particulièrement atypique par rapport à nos partenaires ». Si les « partenaires » sont les Etats-Unis, on peut se réjouir de ne pas connaître en France l'anarchie qui caractérise la distribution du médicament dans ce pays (cf. infra, notamment le jugement de l'attorney general de l'Etat du Michigan et les constatations du GAO en juin 2004) ; si c'est l'Allemagne, c'est faux, tout au moins jusqu'à une date très récente. En effet, nos voisins d'Outre-Rhin sont en train de découvrir une nouvelle organisation de la distribution du médicament où tout devient possible, exploitant les facultés ouvertes par un arrêt de la CJCE qu'il convient précisément d'aborder.
* L'arrêt « DocMorris » rendu le 11 décembre 2003 par la Cour de Justice des Communautés Européennes
Cet arrêt a été rendu en réponse à une question préjudicielle posée par le Landesgericht (tribunal du Land) de Francfort-sur-le-Main dans le cadre d'une procédure engagée par les fédérations et associations de pharmaciens allemands contre la société DocMorris, établie aux Pays-Bas à cinq kilomètres de la frontière germano-néerlandaise. Celle-ci propose à la vente par Internet des médicaments soumis ou non à prescription médicale, notamment vers le public allemand. C'est sur la base de la législation allemande alors en vigueur que cette instance avait été engagée. En effet, cette législation interdisant la vente par correspondance des médicaments dont la délivrance est réservée exclusivement aux pharmacies et qualifiant d'illégale toute publicité tendant à favoriser une telle vente par correspondance, le Deutscher Apothekerverband (DAV) a engagé une procédure contentieuse à l'encontre de DocMorris devant les juridictions allemandes. Le litige s'inscrivant dans un contexte communautaire et transfrontalier, le juge allemand a décidé de surseoir à statuer afin de poser des questions préjudicielles à la CJCE.
Cet arrêt est d'une grande complexité car il fait notamment appel à des notions de droit communautaire subtiles relevant de différents domaines ; il ne saurait donc être analysé en détail ici d'autant que c'est par ses répercussions éventuelles qu'il rentre dans le périmètre de la présente évaluation de la loi de 1998. Mais compte tenu de l'ampleur du phénomène des ventes de médicaments par Internet dans des pays étrangers lointains et quelques uns proches (Pays-Bas, Suisse par exemple), il convient de rappeler les termes de cette évolution inquiétante et donc de cet arrêt.
Une communication de M. Eric Fouassier, professeur à l'Université de Paris XI et membre du Conseil national de l'Ordre des pharmaciens (bulletin de l'Ordre de mars 2004 n° 382) procède à cette analyse approfondie et très argumentée. On en retient donc quelques extraits significatifs.
L'objectif de cette étude est ainsi précisé :
« Cette analyse nous permettra de constater que la CJCE s'est nettement prononcée en faveur du commerce transfrontalier de certains médicaments via Internet, ce qui n'était pas si évident au regard du droit dérivé. Cette position est cependant directement liée à la réaffirmation de la nécessité du monopole pharmaceutique. A bien des égards, le cadre juridique dans lequel devra s'inscrire ce type d'activité demeure à définir. La CJCE n'a pas tranché en effet la question du régime juridique des pharmacies virtuelles ».
M. E. Fouassier enchaîne en donnant ensuite son appréciation préliminaire sur le sens de et arrêt :
« Une simplification abusive du langage, liée à une approche sans nuances des concepts, consisterait à considérer que la CJCE vient d'autoriser la vente des médicaments sur Internet. Se contenter de compléter cette affirmation en précisant que l'autorisation se limite aux seuls médicaments non soumis à prescription serait tout aussi superficiel. Il convient en effet de faire remarquer que cette vente est déjà possible chez certains de nos voisins européens. Le meilleur exemple nous est précisément fourni par la société DocMorris, qui exerce cette activité en toute légalité depuis plusieurs années aux Pays-Bas. C'est seulement parce que cette société a voulu étendre son activité au-delà du territoire batave qu'un litige à vu le jour. Cela nous conduit à préciser que l'arrêt DocMorris porte en réalité, non pas tant sur le principe général de vente de médicaments sur Internet que sur le commerce transfrontalier de médicaments via Internet ».
Un autre auteur (M. Jérôme Peigné, Professeur à l'Université de Lille 2) dans un récent article de la revue de droit sanitaire et social (n° 40 avril-juin 2004 - pages 369-384) procède à un long examen de cet arrêt après un bref signalement ainsi rédigé :
« Au regard des articles 28 et 30 CE, un Etat membre ne saurait interdire la vente par correspondance, via un site Internet, de médicaments légalement autorisés et ne nécessitant pas une prescription médicale préalable. En revanche, compte tenu des exigences liées à la protection de la santé humaine, une législation nationale peut prohiber le commerce électronique de médicaments soumis à prescription médicale obligatoire. De la même manière, la publicité faite en faveur de la vente en ligne de médicaments soumis à prescription médicale facultative ne saurait être interdite au regard des dispositions du code communautaire des médicaments à usage humain, alors qu'une telle interdiction reste permise pour la vente de médicaments nécessitant une prescription médicale ».
Dans l'appréciation de l'environnement juridique fait par M. E. Fouassier, il pointe à diverses reprises le caractère peu convaincant de certains raisonnements faits par la CJCE dans l'appréciation des situations et de la portée de certains principes généraux contenus dans le Traité (art. 28 et 30) et les directives (97/7/CE) concernant la protection des consommateurs et 2000/31/CE relative à certains aspects juridiques des services de la société de l'information.
Se référant à la directive 97/3 (art. 3), il observe ainsi :
« L'article 3 de la directive traduit ces considérations en termes de droit. En effet, tout en posant pour principe que « les Etats membres ne peuvent, pour des raisons relevant du domaine coordonné, restreindre la libre circulation des services de l'information en provenance d'un autre Etat membre », il permet de déroger à cette interdiction lorsque cela apparaît nécessaire pour garantir la protection de la santé publique.
Ce rappel du droit communautaire susceptible de s'appliquer aux pratiques commerciales liées à l'apparition des nouvelles technologies révèle que celui-ci est sous-tendu par la volonté politique d'en écarter la vente de médicaments, ou tout au moins de traiter celle-ci de façon singulière. Le juge communautaire pouvait-il s'en affranchir en se penchant pour la première fois sur la matière, à l'occasion de l'arrêt DocMorris ? On était d'autant moins enclin à le penser que toutes les interventions au cours de l'instruction ont insisté sur la nécessité de réserver un traitement d'exception aux médicaments, en résumé de les exclure du champ du commerce électronique. C'est pourtant une solution inverse qu'a adoptée la CJCE le 11 décembre 2003 ».
Autre critique sur la situation comparée des pharmacies allemandes en l'espèce par rapport à des pharmacies étrangères :
« Le gouvernement allemand reconnaissait que l'AMG (loi sur les médicaments) rendait l'accès au marché allemand plus difficile pour les pharmacies étrangères, mais faisait observer que, du fait de l'obligation d'exercice personnel pesant sur le pharmacien, les pharmacies établies sur le sol national n'avaient pas non plus un accès illimité à l'ensemble du marché allemand. C'est cette analyse que rejette la CJCE.
Selon les termes de l'auteur, « l'argumentation développée par la CJCE ne nous apparaît pas totalement convaincante ». Comme nous l'avons vu plus haut, nous envisageons ici le cas de médicaments qui ont obtenu une AMM valable en Allemagne. Dans la quasi-majorité des cas, ces médicaments seront donc déjà commercialisés de façon traditionnelle en Allemagne par les pharmacies nationales. Ainsi l'interdiction de vente par Internet n'empêche nullement l'accès du marché allemand pour ces produits originaires d'autres Etats-membres, mais pouvant être légalement commercialisés en Allemagne.
C'est cependant ce raisonnement contestable qui permet à la CJCE de considérer que l'interdiction nationale de vente par correspondance des médicaments dont la vente est réservée exclusivement aux pharmacies constitue une mesure d'effet équivalent au sens de l'art. 28 CE ».
Enfin, le commentaire de M. E. Fouassier sur les risques que ferait courir la vente de médicaments par Internet pour ce qui concerne les médicaments non soumis à prescription (dont la CJCE estime précisément dans son arrêt qu'elle ne peut être interdite), à partir des points 113 et 114 de l'arrêt est particulièrement critique :
« La Cour souligne même que « l'achat par Internet pourrait présenter des avantages tels que la possibilité de passer commande à partir de la maison ou du bureau, sans nécessité de déplacement, et de formuler calmement les questions à poser aux pharmaciens, avantages qui doivent être pris en considération (point 113). Concernant les risques de mésusage ou d'abus, la Cour précise qu'un tel risque pourrait être diminué grâce à l'augmentation des éléments interactifs existant sur Internet devant être utilisés par le client avant que celui-ci ne puisse procéder à un achat. S'agissant de la possibilité d'abus, il n'est pas évident que, pour ceux qui souhaitent acquérir de façon abusive des médicaments qui ne sont pas soumis à prescription médicale, l'achat effectué dans des pharmacies traditionnelles présente, en réalité, plus de difficultés que l'achat par Internet (point 114 »).
Sur ce dernier point également, on peut ne pas partager l'approche de la Cour qui procède par simple voie d'affirmation. Le raisonnement nous paraît même un peu contradictoire. La Cour estime en effet que le client dispose, avec Internet, de plus de temps et de calme pour exposer son cas et formuler d'éventuelles questions (élément positif). Mais elle ne perçoit pas que cet avantage s'accompagne aussi d'un inconvénient : le client souhaitant acquérir des médicaments de façon abusive disposera de plus de temps pour répondre aux questions éventuellement gênantes du pharmacien. Ce dernier ne pourra plus prendre en compte un trouble éventuel pour pousser plus loin son interrogatoire. Il est donc, selon nous, erroné de penser que la vente via Internet ne facilite pas les demandes abusives ».
Selon l'un des auteurs précités, M. Jérôme Peigné (op.cit. page 381) « l'arrêt DocMorris devrait obliger certains Etats à modifier leurs droits nationaux pour tenir compte des potentialités ouvertes par Internet » ; il rappelle alors les dispositions applicables actuellement en droit français :
« En droit français, les dispositions de l'article L. 5125-25 du code de la santé publique interdisent aux pharmaciens ou à leurs préposés de solliciter des commandes auprès du public (le colportage). Il leur est également interdit de recevoir des commandes de médicaments et autres produits relevant de leur monopole par l'entremise habituelle de courtiers et de se livrer au trafic et à la distribution à domicile de ces médicaments et produits dont la commande leur serait ainsi parvenue. L'article L. 5125-26 du même code interdit, en outre, la vente au public de tous médicaments et produits réservés au monopole des pharmaciens par l'intermédiaire de maisons de commission, de groupements d'achats ou d'établissements possédés ou administrés par des personnes non titulaires de l'un des diplômes exigés pour exercer la pharmacie.
Le droit interne prohibe donc la sollicitation directe et indirecte de commandes auprès du public, ce qui revient à condamner toute vente par correspondance de médicaments à l'aide d'un site Internet. L'interdiction comporte toutefois des aménagements. L'article L. 5125-32 du code de la santé publique a renvoyé au pouvoir réglementaire le soin de préciser les conditions dans lesquelles pouvaient s'effectuer soit la dispensation par le pharmacien ou l'un de ses préposés d'une commande au domicile des personnes se trouvant dans l'impossibilité de se déplacer, en raison de leur âge, de leur état de santé ou de leur situation géographique (art. R. 5104-4 c. santé publ.), soit la livraison à domicile d'une commande par l'entremise d'un tiers, non pharmacien, sous réserve de respecter certaines conditions de transport et d'empaquetage (art. R. 5104-1 c. santé pub) ».
C'est naturellement l'existence de l'ensemble des règles de protection de la santé publique et des principes dont elles découlent qui constituent un obstacle aux appétits commerciaux d'entreprises comme DocMorris laquelle, avant même que l'arrêt de la CJCE ait été rendu et avant que la législation allemande ait été complètement changée avec l'autorisation de vente de tous les médicaments par Internet (cf.infra), faisait part d'objectifs particulièrement ambitieux comme en atteste un article extrait de la revue « Pharmaceutiques » de novembre 2003 (n° 111).
« DocMorris à la conquête d'autres pays » ?
DocMorris, qui convoite 30 % des parts de marché du secteur médicament en Allemagne, ne dégage pour l'instant aucun profit. Le chiffre d'affaires de 2003 d'environ 45 millions devrait quadrupler avant 2005.
Quand on interroge les responsables de DocMorris sur leurs intentions de conquête d'autres marchés en Europe, et particulièrement en France, la réponse est claire : « DocMorris est une pharmacie néerlandaise, qui répond à toutes les conditions européennes d'exercice de cette profession. Nous n'avons pas besoin d'autorisation spéciale pour vendre des médicaments dans tous ces pays selon le droit européen. C'est aux pays de l'Union de transposer le droit européen dans leurs droits nationaux ! Des clients de toute l'Union européenne commandent déjà des médicaments sur notre site ».
Les procédures, mécanismes et garanties qui assurent actuellement la sécurité dans la dispensation des médicaments en France où, de l'avis général, la situation est tout à fait satisfaisante, notamment par rapport à nos partenaires et voisins, ne sauraient être amoindries, le pharmacien n'apparaissant plus qu'au bas d'une rubrique d'un site internet comme « responsable de l'officine » (localisé éventuellement à des centaines de kilomètres).
L'autre élément clé est constitué par l'existence du remboursement par l'assurance-maladie. Il s'agit là en effet d'une réalité qui, de l'avis général, constitue un obstacle à peu près absolu à la concrétisation du risque internet pour la distribution du médicament en France ; et de citer la situation totalement différente des Etats-Unis où l'on évalue à 40 % le nombre de personnes non couvertes par un régime d'assurance-maladie pour ce type de prestation ; avant même l'apparition d'internet, la situation de la distribution du médicament était déjà problématique dans ce pays.
Si cette constatation est tout à fait fondée, il reste que les « médicaments grand public » non remboursés échappent à ce frein et que, d'autre part, les décisions de moindre ou de non-remboursement peuvent alimenter une tendance favorable aux tentatives de distribution par internet.
Enfin, il conviendra de s'assurer qu'aucun médicament qui aurait été acquis via un réseau Internet n'est remboursé par les caisses d'assurance-maladie. La réalisation d'un risque comme celui d'Internet dans la distribution des médicaments est illustrée d'une manière éclatante avec le développement actuel de la situation en Allemagne.
5.2. La distribution par internet du médicament en Allemagne : un cas d'école
Jusqu'en 2003, le régime de distribution au public du médicament était assez semblable à ce que l'on connaît en France. L'objectif du rétablissement de l'équilibre de l'assurance-maladie avait incité les autorités responsables (gouvernement fédéral, caisses d'assurance-maladie et l'ensemble des partenaires de la prévoyance sociale) à exercer une pression pour réduire le poste important que représentent les médicaments. D'après des appréciations globales, les médicaments ont un prix public sensiblement plus élevé en Allemagne qu'en France ; cette situation particulière n'est donc pas étrangère à la force avec laquelle ces pressions se sont exercées.
5.2.1. Les importations parallèles de médicaments
Depuis 1993 est appliqué en Allemagne un accord-cadre passé entre les associations de caisses d'assurance-maladie et l'association des pharmaciens allemands. Au terme de cet accord, le pharmacien d'officine est tenu de fournir une certaine proportion minimale de ces médicaments importés ; cette part dans le chiffre d'affaires est passée de 5,5 % à 8 % en janvier 2003. Un certain nombre de précautions quant à la qualité du médicament, les notices originales et celle incombant en plus à l'importateur ont été édictées.
5.2.2. D'autres mesures de « libéralisation » du médicament
D'autres mesures, dont l'autorisation de vente par internet, ont été prises dans le cadre de la loi du 14 novembre 2003 de modernisation de l'assurance maladie légale. On peut simplement les citer pour donner la dimension de la motivation financière et expliquer ainsi la rigueur, pour ne pas dire la brutalité, (internet par exemple) de certaines dispositions : participation financière accrue des patients aux dépenses de médicaments, transformation du mode de rémunération des pharmaciens, augmentation (de 6 % à 16 %°) du rabais obligatoire donné par les firmes pharmaceutiques aux caisses d'assurance-maladie, ponction fiscale spécifique, mise en place de « prix de référence » pour les médicaments brevetés.
Par ailleurs, le recours aux médicaments génériques, comme c'est maintenant le cas en France, est fortement encouragé par un intéressement direct des pharmaciens. Enfin, d'autres mesures ont été prises en matière de modulation des prix et/ou des taux de remboursement. Ces mesures s'inscrivent dans une logique de régulation financière du système d'assurance maladie, mais elles sont très loin de ce qui se passe depuis peu avec Internet.
5.2.3. La vente par internet elle-même
L'offensive du Néerlandais DocMorris (cf. supra) sur l'Allemagne a commencé en janvier 2000 en proposant sur son site basé aux Pays-Bas, en allemand notamment, de vendre des médicaments par Internet en toute illégalité. Les pharmaciens allemands ont riposté en poursuivant cette société néerlandaise (cf. supra). Bien avant que l'arrêt de la CJCE saisie par question préjudicielle par le tribunal allemand compétent soit rendu, les caisses d'assurance-maladie ont invité leurs assujettis à commander leurs médicaments par Internet (en toute illégalité donc) et en assortissant cette incitation d'avantages financiers.
Les organisations professionnelles de pharmaciens allemands ont créé en 2001 leur propre site internet de commande qui permettait de commander à l'avance des médicaments qu'il fallait ensuite retirer physiquement dans la pharmacie la plus proche du patient.
* Avant même que la procédure européenne et nationale de l'affaire DocMorris ait été achevée, le gouvernement fédéral allemand a reconnu (art. 23 de la loi du 14 novembre 2003 précitée) aux pharmaciens le droit de vendre par Internet tous les médicaments . Ainsi, la distinction entre les diffirents types de médicaments n'apparaît même pas dans la nouvelle législation allemande : les médicaments sur prescription médicale exclusive et les médicaments n'exigeant pas de prescription, peuvent être désormais vendus par internet.
Il est à noter que, dans ce cas, cela ne paraît pas, aux yeux des commentateurs, contraire à l'arrêt DocMorris puisque si le texte de cette décision « permet aux Etats-membres d'édicter une interdiction de vente par correspondance des médicaments nécessitant une prescription médicale, elle ne les oblige cependant pas à le faire » (J. Peigné op.cit. page 385).
Il se révèle ainsi clairement que cette décision a pour effet de permettre un affranchissement maximal des règles de santé publique dont la sphère réservée est pourtant garantie par les traités (art. 28 et 30 CE), mais qu'elle ne saurait rien imposer pour pérenniser ces règles, même lorsqu'elle en accepte l'existence et souligne leur bien-fondé.
Bien entendu, des mesures prévoyant un « système d'assurance qualité » ont été prévues, sur la base de nouveaux articles introduits dans la loi sur les pharmacies d'officine (Apotekengesetz) ...
Brèche supplémentaire dans l'exercice personnel de la pharmacie, mais contrepartie sans doute destinée à rassurer certains praticiens, la loi du 14 novembre 2003 permet désormais de détenir plus d'une officine : une principale et trois filiales. Mais il apparaîtrait que certains voulaient aller plus loin encore, en demandant la possibilité de constituer des chaînes. Il y a là sans doute un cousinage d'inspiration avec les propositions du rapport Camdessus (cf. supra).
D'après des informations très récentes (Deutsches Ärzteblatt du 19 novembre 2004), la distribution de médicaments par Internet a été très limitée jusqu'à présent (40 millions d'euros). Néanmoins, on peut sans difficulté imaginer ce qui est prévisible. La survenance de quelques problèmes qui prendront d'autant plus d'importance que le flux se développera. Dès 2002, M. Eberhard Sinner, ministre de la santé de Bavière avait déjà signalé l'accroissement du nombre de contrefaçons, en particulier celles touchant des médicaments vendus par Internet. La caisse d'assurance-maladie de Berlin aurait enregistré de son côté d'importantes fraudes sur des médicaments génériques. Cette déréglementation pour ne pas dire cette « déconstruction » des mécanismes de santé publique et de sécurité sanitaire entraîne inévitablement le développement de pratiques illégales, voire criminelles, lorsque les contrefaçons de différents types se répandent, à l'instar de ce que l'on enregistre depuis longtemps déjà dans des pays en voie de développement ou aux Etats-Unis par exemple où Internet constitue un sérieux facteur aggravant.
5.3. Contrefaçons et faux médicaments
* Le constat de l'OMS
Le rappel de la définition que donne l'OMS (Organisation Mondiale de la Santé) du médicament contrefait permet, avant même de préciser l'ampleur du problème, de pointer toutes les facettes de ce phénomène criminel qui a déjà une extension considérable et qui se développe dans des zones géographiques jusque là indemnes :
« Définition de l'OMS du médicament contrefait
Produit présenté de façon délibérément frauduleuse quant à sa source et/ou son identité. Les contrefaçons peuvent concerner aussi bien les médicaments de marque que les génériques. Ils concernent :
- des produits contenant des composant corrects
- ceux contenant des composants incorrects
- ceux sans principes actifs
- ceux contenant des quantités incorrectes de principes actifs
- ceux présentant un conditionnement imité ».
L'OMS recense depuis 1982 les contrefaçons de médicaments mais souligne elle-même l'extrême difficulté de valider et de classer les différents produits et filières d'une manière homogène. Il n'en reste pas moins que certains ordres de grandeur peuvent être retenus. 70 % des cas rapportés concernaient les pays en développement ; moins de 30 % de ces produits provenaient des pays développés ; les pays ayant rapporté des cas représentaient moins de 15 % des membres de l'OMS. Dans le domaine des contrefaçons, mais en fait principalement des faux médicaments, l'Afrique est la plus exposée avec par exemple un taux de 70 % pour les anti-paludéens dans sept pays d'Afrique subsaharienne dont le Cameroun, par exemple. Des pourcentages de l'ordre de 12 % sont couramment avancés pour la Russie, la Chine, 16 % au Vietnam et 8,5 % en Thaïlande.
Sur le plan économique mondial, le préjudice est évalué à 35 milliards de dollars américains, en progression de 50 % depuis 1998. 40 ( * ) Les quelques statistiques qui peuvent être mises en perspective montrent une forte accélération du phénomène au cours des dernières années.
* La situation dans plusieurs pays :
En Afrique
Dans le cas des anti-paludéens contrefaits relevé plus haut (six pays d'Afrique subsaharienne), des analyses de comprimés de chloroquine ont montré que seuls 58 %, de ces produits avaient une teneur acceptable en principe actif et que moins de 25 % donnait des résultats acceptables lors des tests de dissolution. D'après les scientifiques qui ont réalisé ces analyses, la mauvaise qualité de ces comprimés pourrait être l'une des causes des taux élevés de résistance à la chloroquine dans ces pays (cité par M. Philippe Duneton lors du colloque du 3 novembre 2004).
La FDA a confirmé récemment en février 2004 un pourcentage de 50 % de contrefaçons pour les anti-paludéens en Afrique. En Guinée, le président de l'Ordre des pharmaciens (M. Fodé Fofana), réunissant des résultats de thèses de doctorat sur les médicaments proposés sur les marchés de Conakry a relevé un taux de non-conformité de 60 % (soit 83 médicaments) ; 23 % ne comportaient aucune substance active. De nombreuses classes thérapeutiques étaient concernées : antibiotiques, anti-inflammatoires, antiseptiques, antihistaminiques, antitussifs, antiparasitaires et diurétiques.
Le cas du Liban
La situation du Liban relève du cas d'école. Dans un pays où la distribution et la dispensation du médicament étaient organisées sur des bases convenables et efficaces, un changement brutal de législation en mai 2002 autorise la pratique d'escomptes (de ristournes) sur le prix des médicaments ; selon le président de l'Ordre des pharmaciens libanais (M. Ziad Nassour), le but déclaré était de faire baisser le prix des médicaments, mais surtout de créer une diversion au moment de l'entrée en vigueur de la loi relative à la TVA.
Selon M. Nassour, « l'incitation à la consommation a finalement fait exploser la contrebande et l'irruption des contrefaçons » 41 ( * ) ; avec un médicament authentique acheté 80 livres contre un faux ou une contrefaçon achetée 15 livres, les pharmaciens honnêtes ont été rapidement mis dans une situation intenable et des faillites s'en sont suivies. Quant à l'objectif de baisse des prix, l'augmentation de la consommation et les effets désastreux en terme de santé publique permettent de le remettre en perspective, notamment pour ceux qui, en Europe, semblent décidés à orienter les décisions dans ce domaine exclusivement en fonction de cet objectif.
Les États-Unis
-- A partir d'une situation déjà différente de celle que l'on connaît dans la plupart des pays d'Europe occidentale, les Etats-Unis ont enregistré depuis quelques années une dégradation sensible de la sécurité des médicaments dispensés au public. M. Yves Juillet (Les entreprises du médicament) a fait part des informations suivantes lors du colloque précité :
« Les Etats-Unis ont vu doubler les déclarations de cas de fraude entre 2003 et 2004. Il s'agit de produits chers à forte valeur ajoutée (hormones, EPO, anticancéreux), le phénomène est aggravé par la couverture médicale incomplète. 40 millions d'Américains n'ont pas de protection sociale. Et contrairement à la France, le circuit de distribution des médicaments est perméable, les pharmaciens peuvent acheter à qui se prétend distributeur sans vérification ».
De son côté, la FDA dans son rapport publié en 2004 (« Combattre la contrefaçon des médicaments ») signale l'augmentation depuis 2001 des enquêtes sur des médicaments contrefaits ».
-- La distribution par Internet n'est pas la seule en cause dans cette évolution inquiétante, mais elle joue un rôle considérable d'après les Américains.
L'indication qu'en donne M. Eric Fouassier dans son article précité (Bulletin de l'Ordre des pharmaciens n° 382 mars 2004 page 112) mérite d'être citée par « l'ambiance » qu'elle évoque sur la base de réalités avérées :
« Cette prise de conscience d'un danger potentiel pour la santé publique s'est fait sentir même aux Etats-Unis. Dans un contexte de liberté créatrice proche de l'anarchie, des garde-fous se sont mis peu à peu en place pour canaliser des initiatives parfois fort éloignées de l'éthique médicale. La Food and Drug Administration (FDA) s'est attachée à traquer les sites sauvages qui présentaient des allégations mensongères concernant la présence de personnel médical et le contrôle des ordonnances par un pharmacien, ou bien qui livraient des produits prohibés. Des dizaines de pharmacies en ligne ont fait l'objet d'enquêtes pour vente de médicaments de prescription sans ordonnance et vente de produits non autorisés ou illégaux 42 ( * ) . Au début de l'été 2000, ces investigations avaient conduit à la condamnation de 22 personnes et à l'arrestation de 43 autres. L'Etat du Michigan avait purement et simplement interdit la vente de médicaments sur Internet et menacé de poursuites judiciaires les pharmacies en ligne ne respectant pas cette interdiction. L'attorney general de cet Etat avait également assorti sa décision de commentaires peu amènes sur les sociétés de vente de médicaments par e-mail : « C'est un énorme problème. Si maintenant on peut donner des médicaments à n'importe qui et sans ordonnance, autant mettre ces médicaments sur les étagères d'une épicerie 43 ( * ) ».
-- Le rapport du GAO de juin 2004 (n° 04-820)
En juin 2004, le General Accouting Office des Etats-Unis (équivalent de la Cour des Comptes) a rendu un rapport (au Président de la sous-commission permanente des enquêtes de la commission des affaires gouvernementales du Sénat) sur les « pharmacies sur internet ». La synthèse de ce rapport, publiée dans le bulletin « GAO Highlights » est tout à fait conforme à la forte inquiétude que tous les observateurs ont précédemment ressentie.
En voici quelques extraits :
« Les constatations du GAO
Le GAO a pu se procurer, sans aucune prescription, la plupart des médicaments délivrés sur ordonnance qu'il recherchait, auprès de divers sites de pharmacies en ligne. Le GAO a ainsi obtenu 68 échantillons de onze médicaments différents -chacun auprès d'une pharmacie en ligne différente aux États-Unis, au Canada et dans d'autres pays étrangers parmi lesquels l'Argentine, le Costa Rica, les Fidji, l'Inde, le Mexique, le Pakistan, les Philippines, l'Espagne, la Thaïlande et la Turquie. Cinq des pharmacies américaines et la totalité des 18 pharmacies canadiennes, dont le GAO a reçu des échantillons, ont réclamé que le patient présente une ordonnance, tandis que les 24 pharmacies américaines restantes et la totalité des 21 pharmacies étrangères hors Canada ont délivré une ordonnance sur la base de leur propre questionnaire médical, voire n'en ont exigé aucune. Parmi les médicaments que le GAO a obtenus sans prescription figuraient des substances soumises à des restrictions spéciales et des antalgiques à effet narcotique, entraînant une forte accoutumance.
Le GAO a répertorié divers problèmes concernant la manutention, l'autorisation de mise sur le marché de la FDA et l'authenticité des 21 échantillons adressés par les cyberpharmacies étrangères hors Canada. Des problèmes, moins nombreux, ont été constatés parmi les pharmacies du Canada et des États-Unis. Aucune des pharmacies étrangères hors Canada n'exige un étiquetage pharmaceutique fournissant des instructions d'utilisation, peu formulent des mises en garde, et 13 posent des problèmes concernant la manutention des substances. Ainsi, trois échantillons de médicament, qui auraient dû être expédiés en atmosphère thermocontrôlée, sont parvenus dans une simple enveloppe non isolée. Des tests de fabricants ont révélé que la plupart de ces échantillons ne bénéficiaient pas d'autorisation pour le marché des États-Unis ; toutefois, les fabricants ont constaté que la composition chimique de tous ces produits était comparable à celle du produit commandé par le GAO, sauf pour quatre d'entre eux. Quatre échantillons, en effet, se sont révélés être des produits de contrefaçon ou d'une composition ne pouvant être comparée au produit commandé. Comme pour les échantillons reçus des autres pharmacies étrangères, les fabricants ont établi que les échantillons provenant des officines canadiennes étaient, pour la plupart, non autorisés sur le marché des États-Unis, mais qu'en revanche, la composition chimique de tous les échantillons de cette provenance était à chaque fois comparable au produit commandé par le GAO.
(...)
De même, nous avons constaté que certains échantillons avaient été expédiés à partir d'adresses sujettes à caution, notamment à partir de domiciles privés. Nous avons également constaté que certaines cyberpharmacies dissimulent l'information sur le médicament vendu, notamment des pharmacies étrangères hors Canada auprès desquelles nous avions commandé un médicament de marque, mais dont nous avons reçu tantôt le générique, tantôt la version locale. Enfin, il est apparu que 21 pour cent des pharmacies nous ayant fait parvenir des échantillons étaient sous le coup d'une enquête de la DEA ou de la FDA. Ces enquêtes ont été déclenchées parce que les pharmacies concernées étaient soupçonnées de vendre des médicaments frelatés, d'utiliser des désignations commerciales frauduleuses, de pratiquer la contrefaçon, ou encore de vendre des médicaments, normalement délivrés sur prescription, en l'absence de toute relation patient-médecin valable. Neuf de ces pharmacies étaient originaires des États-Unis, une du Canada et cinq d'autres pays étrangers.
Nous avons soumis un projet du présent rapport à la FDA, laquelle s'est montrée en général d'accord avec nos résultats et nos conclusions. Nous avons également soumis un projet du présent rapport à la DEA aux fins d'un examen technique, et celle-ci nous a informés qu'elle n'avait aucun commentaire à formuler.
(...)
Remarques de conclusion
Les consommateurs peuvent obtenir directement par Internet des médicaments, normalement délivrés sur prescription, sans présenter d'ordonnance - en particulier en passant par certaines cyberpharmacies aux États-Unis et des pharmacies étrangères hors Canada. Les médicaments ainsi proposés incluent des substances qui imposent au patient une surveillance en raison de leurs effets secondaires, ou bien qui l'expose à un risque de toxicomanie. S'agissant des médicaments de ce type en particulier, la prescription et la surveillance médicale peuvent contribuer à garantir la sécurité du patient. Outre l'absence de prescription obligatoire, certaines cyberpharmacies peuvent exposer les consommateurs à d'autres risques. Ainsi, notre étude a montré que de nombreuses pharmacies étrangères hors Canada délivrent des médicaments sans fournir d'instructions pour leur utilisation au patient, que les mises en garde sont peu fréquentes et que quatre d'entre elles ont expédié des médicaments, qui n'étaient pas les produits authentiques que nous avions commandés.
On remarquera que nous avons identifié des problèmes somme toute nombreux, en dépit du nombre limité de substances achetées, des problèmes qui correspondent à ceux signalés récemment par les autorités de régulation fédérales et locales ».
En Grande-Bretagne
Depuis 1994, la Grande-Bretagne n'avait pas été confrontée à l'irruption de médicaments contrefaits dans la chaîne pharmaceutique (deux occurrences relevées cette année-là). En août et septembre 2004, deux cas ont été signalés sur deux médicaments différents, l'un d'entre eux ayant été remarqué par le grossiste de lui-même à partir du repérage d'un numéro de lot inhabituel.
Au sujet de ces événements, l'Ordre des pharmaciens français a fait le commentaire suivant (26 novembre 2004) :
« La majeure partie des médicaments contrefaits se retrouvent sur les sites Internet de vente de médicaments. En effet, la chaîne pharmaceutique britannique est très contrôlée et l'apparition de médicaments contrefaits dans la chaîne pharmaceutique britannique est relativement rare (il s'agit du deuxième cas en dix ans). Le potentiel de risque de survenue de contrefaçon dans la chaîne est faible ; la seule faille connue, d'après les experts, est l'importation parallèle de médicaments en provenance d'autres Etats membres de l'Union européenne. Toutefois, les médicaments provenant d'importations parallèles doivent avoir une licence et être clairement identifiés.
Par ailleurs, la majeure partie des Britanniques continue d'acheter ses médicaments à l'officine, suite à la prescription d'un médecin. Les médicaments peuvent ensuite être remboursés par l'assurance maladie (le NHS).
Pour ce qui est des médicaments de « société », les patients ont beaucoup plus tendance à s'auto-médiquer et à rechercher des prix plus intéressants (y compris sur Internet). Or, le Cialis R comme le Réductil R sont tous les deux des médicaments dits de société (ou « life style drugs » en anglais). Ce type de médicaments est donc sujet à de plus nombreuses contrefaçons.
En conclusion, ces deux cas mettent en avant un développement inquiétant de la contrefaçon en Europe ».
On peut rappeler que la distinction que l'arrêt DocMorris de la CJCE s'est crue autorisée à faire entre les médicaments soumis à prescription et ceux qui ne le sont pas montre clairement les risques immédiats que la distribution par internet fait courir.
En France
En France, quelques affaires de contrefaçons de médicaments ou de produits proches ont été trouvées en dehors du circuit pharmaceutique. Les principales affaires de contrefaçons de médicaments ont été signalées par les douanes (Viagra en transit) ou par l'AFSSAPS et la DGCCRF (lentilles optiques). Cela ne veut pas dire que de nombreux sites Internet basés à l'étranger et souvent non identifiables quant à leur localisation et leur nature réelle ne proposent pas ouvertement en France des médicaments. Les assurances que l'on pense avoir avec le remboursement par l'assurance-maladie sont réelles et sans doute pérennes ; en ce qui concerne les produits non remboursables, c'est autre chose. Dans son article précité, M. Eric Fouassier (page 117) indique ainsi (mars 2004) :
« Il existe déjà sur Internet des centaines de sites où l'on peut se procurer des substances de toutes sortes : de l'ecstasy, des psychotropes, des hormones de croissance ou d'autres produits classés sur la liste I des substances vénéneuses. A Montpellier, le 23 mars 1999, huit cents médecins et pharmaciens ont été entendus à titre de témoins dans le cadre d'une information judiciaire sur la vente, via Internet, de médicaments interdits. Selon la plainte du Conseil de l'Ordre des médecins des Pyrénées orientales, ces médicaments censés favoriser l'amaigrissement auraient été vendus par une société installée à Barcelone à des praticiens, dont au moins trois cents exerçaient dans les Pyrénées orientales, l'Aveyron et l'Hérault. La société espagnole approvisionnait les médecins et les pharmaciens grâce à une entreprise installée à Montpellier ».
Pour ceux qui en auraient douté, il y a donc en France, comme ailleurs, et malgré les dispositions légales organisant la dispensation du médicament, des médecins et pharmaciens qui n'hésitent pas à se lancer dans des pratiques frauduleuses et vraisemblablement dangereuses. Il n'y a aucune raison objective à remettre en cause l'organisation actuelle. Les responsables de tous ordres doivent être au contraire vigilants pour ne donner aucune chance à de douteuses entreprises cybernétiques où la sécurité sanitaire et la santé publique ont tout à perdre ; il s'agit là de grands risques humains et financiers pour la collectivité comme pour les personnes.
RESUME DE LA TROISIEME PARTIE
|
* Une structure aux missions étendues Succédant à l'Agence du médicament, l'AFSSAPS a recueilli, par la loi de 1998, un ensemble de compétences beaucoup plus large que cette dernière (extension aux dispositifs médicaux, cosmétiques notamment). Des moyens importants ont été consacrés à la mise en place de la nouvelle structure. Il est à noter qu'au financement budgétaire s'ajoute celui des redevances acquittées par les industriels ; cette dernière ressource ne saurait se substituer à la première car l'Etat a le devoir d'assumer ses responsabilités, en particulier à hauteur de toutes les fonctions de contrôle et d'inspection. * Une montée en puissance difficile Malgré les moyens importants dont elle a bénéficié, l'Agence a connu un démarrage lent et laborieux, tout en continuant à assurer d'une manière satisfaisante, le fonctionnement des procédures d'AMM (autorisation de mise sur le marché) tant nationales qu'européennes qui constituent son « coeur de métier ». Les insuffisances s'expliquent par l'héritage souvent désordonné de l'Agence du médicament avec de sérieuses conséquences sur l'informatique, donc sur les bases de données, essentielles dans ce domaine (information du corps médical, fichier d'experts) avec une gestion interne insuffisante qui a perduré ; l'absence de COM (contrat d'objectifs et de moyens) illustre bien ces faiblesses. Au-delà des responsabilités de l'Agence elle-même, des éléments extérieurs ont été à l'origine d'autres difficultés : retards importants dans l'élaboration des textes d'application de la loi de 1998, exercice de la tutelle du ministère de la santé (DGS notamment) mal détaché de l'ancienne architecture administrative. * Une stabilisation sujette à des remises en cause Maintenant que la stabilisation paraît enfin acquise, plusieurs facteurs peuvent être identifiés qui risquent de la remettre en cause. ? La croissance des activités due aux circonstances fournit toute une série de facteurs en ce sens : -- veille sanitaire pour les médicaments avec des signalements multipliés par 3,5 entre 1999 et 2003 et on imagine l'effet des tempêtes des dernières semaines dans ce domaine ! -- veille face aux risques terroristes (plans Biotox et Piratox) ; -- croissance des besoins dans le domaine des vaccins (libération de lots, contrôle, coopération) ; -- entrée en vigueur du nouveau régime des essais cliniques. En outre, dans tous les secteurs la mobilisation due à la dimension européenne est très forte. ? Les modifications institutionnelles touchant le périmètre des compétences constituent la deuxième série de facteurs : création du FOPIM (Fonds de promotion et d'information médicale et médico-économique) en 2001, suivie de la réforme de la commission de la transparence en septembre 2003, et enfin transfert de l'ensemble de cette dernière au sein de la nouvelle HAS (Haute Autorité de Santé) créée par la loi du 13 août 2004, avec suppression corrélative du FOPIM et intégration de l'ANAES (Agence nationale d'accréditation et d'évaluation en santé). Une actualité lourde d'interrogations Au-delà de l'adéquation des mécanismes de la loi de 1998 se pose la question fondamentale que la crise du Vioxx vient d'éclairer d'une manière encore plus crue que les précédentes. -- La iatrogénie médicamenteuse avait déjà été repérée depuis quelque temps, notamment aux Etats-Unis et dans d'autres pays dont la France. -- Quelques crises récentes ont soulevé de lourdes interrogations finissant par mettre en cause, au-delà d'un médicament ou d'une classe thérapeutique donnée, les modalités d'expertise et les procédures d'AMM elles-mêmes. La cérivastatine retirée du marché mondial par Bayer en août 2001 a été la première de ces crises. La mise en cause des THS (traitement hormonal substitutif) a constitué un autre type de crise où le facteur de la demande sociale joue un rôle important, les produits étant toujours sur le marché alors que les suspicions de danger substantiel se confirment encore tout récemment. Le retrait du Vioxx du 30 septembre 2004 dans des conditions qui rappellent celles de la cérivastatine, constitue un véritable séisme dans le secteur de l'industrie pharmaceutique et même de l'ensemble du monde médical y compris les experts et les agences du médicament. Des doutes sérieux sur les bénéfices additionnels (moindres effets indésirables) par rapport aux anti-inflammatoires préexistants étaient déjà largement répandus. La révélation de risques accrus d'accidents cardiovasculaires notamment, a entraîné le producteur, Merck, à retirer brutalement le Vioxx du marché. Les doutes se sont rapidement répandus sur la date à laquelle ces certitudes avaient été acquises et sur une éventuelle dissimulation par l'industriel. Plusieurs études, dont l'une réalisée en Suisse et publiée début novembre 2004, ont confirmé la disponibilité ancienne de ces informations. L'AFSSAPS avait demandé à l'Agence européenne (EAMA) une réévaluation de cette classe thérapeutique (les coxibs) qui n'a pas été réalisée, semble-t-il, dans les conditions souhaitées. Cependant, tout en renforçant ses réserves, elle avait elle-même maintenu le Vioxx et les autres coxibs sur le marché, comme toutes les agences, y compris la FDA. Les conditions de marketing dans lesquelles le Vioxx a été lancé (prix d'appel très bas en milieu hospitalier et demande de prix public remboursable excessivement élevé) ont contribué à ternir l'image des industriels. Devant les mises en cause pour le moins justifiées que cette crise entraîne, les firmes pharmaceutiques au niveau mondial viennent de s'engager (6 janvier 1995) enfin à faire connaître les résultats de tous les essais cliniques qu'elles réalisent. Il revient aux agences (AFSSAPS et EAMA notamment) et à l'Etat de s'assurer de la pérennité de ces engagements. Ces événements appellent le renforcement substantiel des mécanismes de sécurité, notamment en matière de pharmacovigilance. Ils exigent aussi que l'expertise soit radicalement revisitée, même si une partie des réponses se trouve au niveau mondial, d'autant que certains problèmes rencontrés avec le médicament apparaissent, avec d'autres spécificités, dans d'autres domaines : alimentaire (toxicologie), produits chimiques ... Pour que le « vivier » d'experts soit partout suffisant en quantité et en qualité, des efforts en terme de moyens et de rémunérations s'imposent ; ils sont la condition de leur indépendance, évidemment essentielle à leur mission. Le statut de l'expertise au sens professionnel, intellectuel et social doit lui aussi être substantiellement réévalué, notamment dans le milieu universitaire. Au total, les incertitudes du fonctionnement actuel de l'expertise scientifique justifient l'ouverture d'une large réflexion en vue de la création d'une Haute Autorité de l'Expertise Scientifique pour assurer une indépendance réelle de l'expertise. Les risques émergents -- Dans le cadre européen, un risque nouveau doit être envisagé ; c'est celui de la concurrence entre agences des Etats-membres : les critères de souplesse et de rapidité paraissent pour le moins antinomiques avec la sécurité et la validité dans une procédure d'AMM. Par le jeu de la procédure de reconnaissance mutuelle, il y a là des risques non négligeables d'affaiblissement de la sécurité, en particulier dans une Europe à 25. L'homogénéité de la qualité des mécanismes d'inspection entre Etats-membres soulève des inquiétudes qui ont déjà été relevées à différents niveaux. La « nouvelle approche » de certification européenne pour les dispositifs médicaux peut susciter elle aussi des interrogations justifiées. -- La banalisation du médicament constitue un risque particulièrement grave qui s'inscrit à contre-courant du mouvement de renforcement de la sécurité sanitaire. La France bénéficie d'un système de distribution et de dispensation du médicament qui est un des meilleurs du monde, mais différentes initiatives en Europe et dans le monde fournissent des exemples particulièrement inquiétants avec la vente de médicaments par internet. Le cas des Etats-Unis, où les conditions de distribution du médicament n'étaient déjà pas satisfaisantes, ce qui explique sans doute la iatrogénie qu'on y relève, a amené les autorités américaines à s'inquiéter officiellement des conséquences de la vente par internet : une investigation du GAO (General Accountant Office, équivalent de la Cour des Comptes) réalisée en juin 2004 les précise clairement.. Le cas de l'Allemagne se révèle encore plus inquiétant du fait bien sûr du voisinage et de l'appartenance à l'Union européenne. Jusqu'à la fin de l'année 2003, le système de dispensation du médicament y était proche du système français et ne posait guère de problème de sécurité. Souhaitant sans doute anticiper une décision de la CJCE (Cour de justice des Communautés européennes, Doc Morris - 11 décembre 2003), mais surtout « faire des économies » sur le médicament pour réduire le déficit de l'assurance-maladie, le gouvernement allemand a fait adopter un mois plus tôt dans une loi, la possibilité d'acheter par internet tous les médicaments, y compris ceux soumis à prescription obligatoire. Il allait ainsi très au-delà de ce que la CJCE a autorisé. Les conséquences néfastes d'une déréglementation qui permet la vente par correspondance, et même y incite (par le biais des caisses d'assurance maladie) commencent à se faire sentir. L'encouragement fatal de la contrefaçon et des faux médicaments se vérifie. Toutes les mesures de précaution élaborées par la loi pour éviter débordements de consommation, non respect des contre-indications, effets croisées de différents médicaments, substitution de personnes sont ainsi balayées. On peut se demander si la recherche d'une sécurité sanitaire des produits de santé a encore un sens dans un tel paysage. |
QUATRIÈME
PARTIE :
UNE CLARIFICATION NÉCESSAIRE
I. UNE COMPLEXITÉ CROISSANTE
La loi du 1 er juillet 1998 a eu pour objectif de renforcer la sécurité sanitaire en créant un ensemble efficace et cohérent. L'architecture administrative n'est pas une fin en soi, mais l'éparpillement, les conflits de compétences, la complexité sans justification restent à éviter. Les bases essentielles d'un nouveau dispositif d'ensemble ont donc été posées et celui-ci a commencé à fonctionner dans des conditions indiscutablement satisfaisantes, notamment par rapport au passé. Il reste que la simplification est insuffisante et que des ajouts ultérieurs ont très rapidement apporté des éléments de complication supplémentaires.
1.1. Une simplification incomplète
Dans l'analyse de la situation de départ, la multiplication des structures avait été clairement notée comme l'un des nombreux facteurs de dysfonctionnement de la sécurité sanitaire (cf. supra 1 ère partie). M. Claude Huriet déclarait par exemple lors de la présentation de son rapport sur la proposition de loi à l'automne 1997 : « Il faut mettre un terme à la dispersion des moyens, cesser de confier à de multiples organismes le soin d'accomplir des tâches souvent identiques, nécessitant de recourir à des procédures complexes ».
La création de l'AFSSA, de l'AFSSAPS et de l'InVS a indiscutablement contribué à se rapprocher de cet objectif, mais partiellement. La direction retenue est la bonne, mais la complexité demeurait excessive dès le départ. Le législateur de 1998 n'avait d'ailleurs pas ignoré cet état de fait puisqu'il avait prévu par l'article 3 de la loi : « Dans un délai d'un an suivant la promulgation, le Gouvernement remet au Parlement un rapport ayant pour objet de proposer la restructuration des organismes de droit public propre à éviter une confusion des missions et la dispersion des moyens de veille sanitaire ». Cette disposition à la fois révélatrice et lucide n'a pas été appliquée. On peut le regretter d'autant plus qu'un tel rapport aurait permis d'éviter des décisions intervenues ultérieurement et ont compliqué encore davantage certaines situations. La loi du 9 mai 2001 a elle-même contribué directement à cette évolution. Sans entrer dans le détail de ces situations qui a été abordé les trois précédentes parties du rapport, on peut rappeler l'exemple que donnait précisément M. François Autain, Sénateur, un an après la mise en application de la loi de 1998 lors de la journée d'audition de la commission des affaires sociales du Sénat le 25 mai 2000 (rapport précité page 34) :
« Je me suis amusé à relever le nombre d'instances d'expertise qui existaient dans votre domaine, Monsieur Hirsch, et j'en ai dénombré dix :
- le Conseil Supérieur d'Hygiène Publique,
- la Commission d'Etude des Produits destinés à une Alimentation Particulière
- la Commission Interministérielle et Professionnelle de l'Alimentation Animale,
- la Commission de Technologie Alimentaire,
- le Centre National d'Etudes et de Recommandations sur la Nutrition et l'Alimentation,
- l'Observatoire des Consommations Alimentaires,
- le Visa Préalable de Publicité
- l'Académie de Médecine,
- le Conseil National de l'Alimentation,
- le Conseil National de la Consommation ».
La tendance bien « hexagonale » à maintenir des commissions techniques et échelons spécifiques trouve ici une illustration éclatante alors que l'on souhaite pourtant réellement procéder à une réforme d'ensemble. Des progrès indiscutables ont été réalisés dans le secteur des produits de santé où le mouvement de regroupement a été réel ; il n'en reste pas moins qu'ici aussi, des articulations discutables ont coexisté, voire ont été accrues.
Une remarque s'impose au passage : il ne s'agit pas d'unifier d'une manière artificielle la structure des agences sur un modèle unique ; chaque sphère a sa propre logique avec des principes et des mécanismes qui peuvent, voire doivent, être différents ; mais il s'agit d'éviter que des « strates » de créations administratives antérieures ou latérales puissent perdurer, entamant ainsi l'efficacité de ces mécanismes.
• L'équilibre de l'architecture retenue
en 1998 peut aussi être mis en cause par l'insuffisance de l'application
de la loi elle-même. Cette faiblesse est illustrée par les
produits phytosanitaires qui sont clairement dans la compétence de
l'AFSSA (art. L. 1323-1 du CSP), mais pour lesquels la configuration
administrative antérieure a été maintenue, la loi
étant simplement
« négligée »
sur ce point.
* Les organismes consultatifs et autres
Anciens ou récents, les organes consultatifs dans la sphère de la santé publique et de la sécurité sanitaire ont, par leur nombre et l'enchevêtrement des compétences, contribué eux aussi à rendre encore plus complexe un paysage institutionnel qui l'était déjà passablement. Le champ de la santé n'est pas le seul à connaître ce foisonnement d'organes, mais c'est un de ceux où ce phénomène est le plus marqué alors que précisément les nombreuses réformes législatives et auraient du permettre une rationalisation minimale.
-- Parmi les organes « classiques » se trouve en premier lieu le Conseil Supérieur d'hygiène public de France (CSHPF) . Créé en 1902 (cf. supra 1 ère partie), il a le profil habituel de « conseil supérieur », mais il est resté compétent après 1998 pour émettre des avis ou recommandations et éventuellement réaliser des missions d'expertises spécialement de précision, d'évaluation et de gestion des risques pour la santé de l'homme. Ses membres sont répartis en quatre sections : prophylaxie des maladies transmissibles, eaux, risques de l'environnement sur la santé, alimentation et nutrition. Son secrétariat est assuré par la DGS, ce qui n'est pas sans signification compte tenu de la communauté de compétences évidente avec l'InVS, l'AFSSE, l'AFSSA et d'autres organismes. Ce qui était logique dans une structure ministérielle classique ne l'est plus du tout avec le nouvel ordonnancement institutionnel. Les positions qu'il est amené à prendre pour certaines d'entre elles ont un caractère quasi décisionnaire. La loi de santé publique a logiquement supprimé le CSHPF en transférant l'essentiel de ses compétences au nouveau Haut Conseil de la santé publique, quelques difficultés subsistant encore cependant.
-- Le Haut Comité de la santé publique institué par le décret du 3 décembre 1991 avait pour mission de contribuer à la définition des objectifs de la santé publique, de promouvoir la prévention et de développer l'observation de l'état de la population. La loi du 4 mars 2002 (relative aux droits des malades et à la qualité du système de santé) l'a remplacé par le Haut Conseil de la santé chargé de contribuer à la définition des priorités pluriannuelles de santé publique ; il formule toute recommandation en vue d'améliorer les politiques de santé et évalue dans un rapport annuel leur application.
Par la loi du 9 août 2004, ce Haut Conseil de la santé devient, par fusion avec le CSHPF, le (nouveau) Haut Conseil de santé publique (cf. infra).
-- On ne s'attardera pas sur de nombreux autres organes à caractère consultatif, notamment ceux dont les compétences s'exercent plutôt sous l'angle de l'assurance maladie (conférence nationale de la santé), qui étaient antérieurs à la réforme de la loi sur l'assurance maladie du 13 août 2004.
-- Deux organismes enfin méritent d'être cités pour leur rôle opérationnel apprécié, mais dont le positionnement montre les difficultés d'ordonnancement du paysage institutionnel. Il s'agit de l'INPES et de la Commission de sécurité des consommateurs.
• L'Institut national de prévention et
d'éducation pour la santé (INPES) a succédé en 2002
(loi du 4 mars) au Comité français d'éducation pour
la santé. C'est un établissement public placé sous la
tutelle du ministre de la Santé ; il est chargé d'une
« fonction d'expertise et de conseil en matière de
prévention et de promotion de la santé » et d'une
mission de « développement de l'éducation pour la
santé » (art. L.1417-4, CSP).
En outre, il oeuvre directement pour le développement de l'éducation pour la santé, notamment dans le milieu scolaire. Son effectif s'élève à 110 personnes, majoritairement des cadres. On a là l'exemple d'un organisme de caractère mixte de par ses fonctions dont certaines se trouvent nécessairement dans un domaine commun avec les administrations ministérielles et l'AFSSA.
• La Commission de sécurité des
consommateurs créée par la loi du 21 juillet 1983
contribue dans un domaine tout à fait différent à
appréhender des risques très variés. Avec des moyens
limités malgré un bon fonctionnement, elle ne peut couvrir
l'ensemble des risques qui relèvent théoriquement de sa
sphère. On a là une illustration des « zones
blanches » que le dispositif institutionnel laisse malgré son
foisonnement.
1.2. Une complexité ajoutée
La loi du 9 mai 2001 créant l'AFSSE et l'IRSN devait compléter et simplifier l'architecture de l'ensemble du dispositif de sécurité sanitaire. Elle a plutôt apporté des éléments de complexité supplémentaires.
• Ainsi l'AFSSE est compétente pour
l'évaluation des risques des produits biocides, mais l'InVS l'est aussi
ainsi que la DGCCRF sous certains rapports ; cette nouvelle agence
(l'AFSSE) peut également donner un avis pour ce qui concerne l'impact
des produits phytosanitaires sur l'environnement. Elle a d'ailleurs
été saisie en mars 2004, parallèlement à l'AFSSA,
sur les troubles enregistrés chez les abeilles (cf. supra partie AFSSA).
Dans un autre domaine, celui de l'eau de boisson, l'AFSSA est
compétente, mais l'AFSSE et, jusqu'à la loi du
9 août 2004 (santé publique), le Conseil
supérieur d'hygiène public de France conservait un droit de
regard.
Le problème d'ensemble du positionnement de l'AFSSE fait l'objet d'une analyse et de propositions à la fin de la présente partie. Notons ici simplement que la recherche de cohérence et d'efficacité a été perdue de vue avec la création d'une agence aux compétences potentielles très larges et aux moyens des plus réduits, sans rapport avec les objectifs fixés. D'autre part, ses compétences comportent de nombreux interfaces non rationalisées avec la plupart des organismes préexistants. Là encore, le législateur avait prévu (art. 4) dans les deux ans de l'entrée en vigueur un « rapport sur la rationalisation du système d'expertise dans son domaine de compétence » ; était-ce une preuve de lucidité, de candeur ou l'aveu d'un échec programmé issu d'un compromis boiteux ?
• Dans le secteur des médicaments et plus
spécialement de l'information thérapeutique à destination
des médecins, la création du FOPIM s'est faite en 2001 dans le
cadre d'une loi de financement de la sécurité sociale. Ici aussi
l'examen de cet épisode (cf. supra partie AFSSAPS) donne l'impression
que l'échec était programmé pour de multiples raisons et
que cette création non intégrée dans le dispositif de la
loi de 1998 était de toute façon un élément de
perturbation. Dans ce même secteur précisément, la
création de l'ANAES (Agence nationale d'accréditation et
d'évaluation en santé) en 1996 constituait déjà un
précédent certes intéressant, mais dont
l'intégration n'allait pas de soi. La loi du
13 août 2004 en a prévu la disparition au sein de la HAS
(Haute Autorité de Santé). La clarification ne paraît pas
évidente : le mélange des genres risque d'être
pérennisé, voire aggravé.
Dans le domaine des organes consultatifs, on a observé que la loi du 4 mars 2002 avait transformé le Haut comité de la santé publique en un Haut conseil de la santé.
Au-delà de ces éléments de complexité qui ont été maintenus, voire aggravés, il apparaît que des efforts pour maîtriser cette complexité ont été engagés, notamment à la suite de ce que la crise de la canicule 2003 a révélé, mais que cela ne paraît pour autant susceptible de résoudre toutes les difficultés, anciennes qui perdurent ou nouvelles.
II. DES TENTATIVES POUR MAÎTRISER LES INSUFFISANCES
La complexité qui débouche sur les dysfonctionnements révélés par la crise de la canicule a entraîné des réactions d'ordre concret dans le domaine politique et administratif : détermination des compétences du ministre de la santé en cas de crise, confirmation et extension des pouvoirs de l'InVS, réforme de la transmission des certificats de décès. Un certain nombre d'organes consultatifs ont été réformés également dans le cadre de la loi de santé publique (du 9 août 2004) et des échelons opérationnels ont été restructurés au sein de l'administration du ministère de la santé. Certaines de ces mesures sont positives et paraissent de nature à régler des difficultés avérées ; d'autres dessinent des perspectives qui suscitent davantage de perplexité.
2.1. Des initiatives plutôt positives
Alors que le projet de loi relatif à la politique de santé publique devait être examiné en première lecture par l'Assemblée nationale au début du mois d'octobre 2003, le ministre de la santé, M. Jean-François Mattei a présenté les amendements au projet originel pour concrétiser les mesures qui constituaient une réponse à la crise de la canicule, pour ce qui relevait de la sphère du ministère de la santé au sens large (InVS, y compris les systèmes de recueil de statistiques etc...). Des mesures concrètes avaient déjà été prises au niveau réglementaire sur la transmission des informations relatives aux décès.
* Les nouveaux pouvoirs confirmés du ministre de la santé sont affirmés et concrétisés dans un nouveau chapitre du code de la santé publique intitulé « menace sanitaire grave » comportant cinq articles dont le premier fixe cette nouvelle prérogative du Ministre :
« Menace sanitaire grave : art. L. 3110-1 - En cas de menace sanitaire grave appelant des mesures d'urgence, notamment en cas de menace d'épidémie, le ministre chargé de la santé peut, par arrêté motivé, prescrire dans l'intérêt de la santé publique toute mesure proportionnée aux risques courus et approprié aux circonstances de temps et de lieu afin de lutter contre la propagation de maladies (...) ».
* Les missions de l'InVS ont de leur côté fait l'objet d'une définition et d'une articulation à la fois plus large et plus précise, voire exhaustive. La rédaction précédente de la loi du 1 er juillet 1998 ne laissait déjà aucun doute quant à la réalité de la mission de l'InVS dans une situation telle que celle de la canicule de 2003.
|
Loi du 1 er juillet 1998 |
Loi du 9 août 2004 |
|
« art. L. 1413-2 - Un Institut de veille sanitaire, établissement public de l'Etat, placé sous la tutelle du ministre chargé de la santé, est chargé : 1°D'effectuer la surveillance et l'observation permanente de l'état de santé de la population, en s'appuyant notamment sur ces correspondants publics et privés, participant à un réseau national de santé publique, dans le but : - de participer au recueil et au traitement des données sur l'état de santé de la population à des fins épidémiologiques ; - de rassembler, analyser et actualiser les connaissances sur les risques sanitaires, leurs causes et leurs évolutions ; - de détecter tout événement modifiant ou susceptible d'altérer l'état de santé de la population ; 2°D'alerter les pouvoirs publics, notamment l'Agence française de sécurité sanitaire des produits de santé mentionnée à l'article L. 5311-1, l'Agence française de sécurité sanitaire des aliments mentionnée à l'article L.1323-1 et l'Agence française de sécurité sanitaire environnementale mentionnée à l'article L.1335-3-1, en cas de menace pour la santé publique, quelle qu'en soit l'origine, et de leur recommander toute mesure ou action appropriée ; 3 °De mener à bien toute action nécessaire pour identifier les causes d'une modification de l'état de santé de la population, notamment en situation d'urgence ». |
« art. L. 1413-2 - Un Institut de veille sanitaire, établissement public de l'Etat, placé sous la tutelle du ministre chargé de la santé, a pour missions : 1°La surveillance et l'observation permanentes de l'état de santé de la population. A ce titre, il participe au recueil et au traitement de données sur l'état de santé de la population à des fins épidémiologiques, en s'appuyant notamment sur des correspondants publics et privés faisant partie d'un réseau national de santé publique ; 2°La veille et la vigilance sanitaires. A ce titre, l'institut est chargé : « a) De rassembler, analyser et actualiser les connaissances sur les risques sanitaires, leurs causes et leur évolution : « b) De détecter de manière prospective les facteurs de risque susceptibles de modifier ou d'altérer la santé de la population ou de certaines de ses composantes, de manière soudaine ou diffuse ; « c) D'étudier et de répertorier, pour chaque type de risque, les populations les plus fragiles ou menacées. « Il peut également assurer des fonctions de veille sanitaire pour la Communauté européenne, des organisations internationales et des pays tiers, avec l'accord du ministre chargé de la santé ; « 3°L'alerte sanitaire. L'institut informe sans délai le ministre chargé de la santé en cas de menace pour la santé de la population ou de certaines de ses composantes, quelle qu'en soit l'origine, et il lui recommande toute mesure ou action appropriée pour prévenir la réalisation ou atténuer l'impact de cette menace ; « 4°Une contribution à la gestion des situations de crise sanitaire. A ce titre, l'institut propose aux pouvoirs publics toute mesure ou action nécessaire. « L'institut participe, dans le cadre de ses missions, à l'action européenne et internationale de la France, et notamment à des réseaux internationaux de santé publique ». |
Pour être sûr de ne rien oublier, l'article suivant, consacré aux travaux s'imposant dans le cadre des missions, dispose notamment :
« Art. L.1413-3 du CSP. En vue de l'accomplissement de ses missions, l'InVS :
1°Effectue, dans son domaine de compétence, toutes études, recherches, actions de formation ou d'information ;
2°Met en place les systèmes d'information lui permettant d'utiliser, dans les meilleurs délais, les données scientifiques, climatiques, sanitaires, démographiques et sociales, notamment en matière de morbidité et de mortalité, qui sont nécessaires à l'exercice de ses missions ;
3°Elabore des indicateurs d'alerte qui permettent aux pouvoirs publics d'engager des actions de prévention précoce en cas de menace sanitaire et des actions de gestion des crises sanitaires déclarées ; » (...)
L'accent mis fortement sur les sytèmes d'information, les différentes catégories de données et leur transmission immédiate au ministre vise sans ambages à empêcher la réédition d'une crise du type de celle de 2003.
Ce nouvel ancrage législatif ne pourra pas nuire à l'InVS pas plus qu'aux prérogatives du ministre qui sont ainsi pour tous les deux fixées dans le code en parfaite clarté.
* La situation est beaucoup moins nette pour ce qui concerne la fusion du CSHPF (Conseil supérieur d'hygiène publique) de France et du Haut Conseil de la santé (loi du 4 mars 2002) pour former le Haut conseil de santé publique (loi du 9 août 2004).
L'histoire et le rôle de chacune des deux instances « fusionnées » ayant été évoqués précédemment, on se limitera à indiquer l'intérêt éventuel de cette mesure mais aussi les questions qu'elle soulève.
La fusion de ces deux instances a pour objectif une dynamique résultant de ce regroupement entre un CSHPF qui avait nécessairement perdu une partie de ses attributions avec la création des agences sanitaires en 1998 et un Haut conseil de la santé au parcours et à l'apport incertains. Or les deux instances ont un rôle assez différent et pas nécessairement complémentaire pour autant. La rédaction finale retenue par l'Assemblée nationale en deuxième lecture suite aux remarques du Sénat sur le texte initial du gouvernement a permis d'éviter une confusion certaine. En effet, le transfert intégral des compétences du CSHPF au nouveau Haut Conseil de la santé publique risquait de conférer à celui-ci une hétérogénéité fâcheuse et notamment une interpénétration de ses compétences et celles des agences sanitaires.
Le nouveau Haut conseil de la santé publique a donc pour mission (art. L.1411-4 du CSP) :
« 1° De contribuer à la définition des objectifs pluriannuels de santé publique, notamment en établissant le rapport mentionné à l'article L. 1411-2, d'évaluer la réalisation des objectifs nationaux de santé publique et de contribuer au suivi annuel de la mise en oeuvre de la loi prévue à l'article L. 1411-2 .
« 2° De fournir aux pouvoirs publics, en liaison avec les agences sanitaires, l'expertise nécessaire à la gestion des risques sanitaires ainsi qu'à la conception et à l'évaluation des politiques et stratégies de prévention et de sécurité sanitaire ;
« 3° De fournir aux pouvoirs publics des réflexions prospectives et des conseils sur les questions de santé publique ».
(...)
On note que tout risque d'interpénétration avec les tâches des agences n'est pas vraiment écarté même si le rapporteur du projet à l'Assemblée nationale a précisé que les missions de ce Haut conseil portent « plutôt sur la conception et la gestion des politiques que sur l'évaluation et la gestion des risques, cette dernière expertise étant plutôt du ressort des agences ». Par ailleurs, le transfert des pouvoirs hérités du CSHPF à ce nouveau Haut Conseil comporte bien des pouvoirs consultatifs en matière de règles générales d'hygiène (44 ( * )) .
A ces modifications, dont l'objectif est celui d'une clarification et d'une unification, s'en ajoute d'autres dont le principe est moins clair, mais la perplexité qu'elles inspirent est nettement plus forte.
2.2. Des perspectives qui interrogent
2.2.1. Le Comité national de santé publique (CNSP)
L'inadéquation du CNSS (Comité national de sécurité sanitaire) à ses missions était déjà évidente et largement constatée avant même la crise de la canicule. Il était logique qu'il n'y survive pas. L'instance qui a été imaginée pour le remplacer par la loi de santé publique du 9 août 2004 est le Comité national de santé publique (CNSP) (art. L.1413-1 du CSP).
Aux termes de cet article, le CNSP a pour mission :
« 1°De coordonner l'action des différents départements ministériels en matière de sécurité sanitaire et de prévention ;
« 2°D'analyser les événements susceptibles d'affecter la santé de la population ;
« 3°De contribuer à l'élaboration de la politique du Gouvernement dans les domaines de la sécurité sanitaire et de la prévention et d'en examiner les conditions de financement ».
(...)
Le CNSP est destiné à coordonner de manière permanente les nombreux ministères concernés par la politique de santé publique. Il est exact qu'aucune instance ministérielle chargée explicitement de cette mission n'existait jusque là. Il doit comporter deux sections : l'une chargée de la prévention générale vis-à-vis de maladies ou problèmes largement répandus dans la population (maladies cardiovasculaires, obésité, nutrition), l'autre chargée de la sécurité sanitaire qui était censée être l'objectif principal du défunt CNSS.
D'après les indications qui ont été données à votre rapporteur, le CNSP dans sa fonction interministérielle de sécurité sanitaire sera une instance de concertation et de coordination des ministères impliqués dans la gestion des risques. C'est là que pourra se débattre une véritable politique de sécurité sanitaire, ce qui est plus large que la coordination scientifique des agences. Il s'agira :
-- de définir et partager les objectifs et les priorités de sécurité sanitaire ;
-- de faire émerger une doctrine cohérente de gestion des risques entre les différents secteurs (air, eau, sols ...) ;
-- de veiller à une allocation des ressources cohérentes au regard des priorités ;
-- de coordonner les contrats d'objectifs des différentes agences ;
-- de discuter des modalités de suivi du travail des agences ;
-- de préparer les positions européennes à prendre sur les grandes questions de sécurité sanitaire ;
-- de prévoir les modalités d'évaluation de la politique de sécurité sanitaire ;
-- de développer une gestion prospective et anticipatrice.
En outre, il est envisagé que le CNSP soit appelé à reprendre les échanges qui se déroulaient au sein du CNSS. Cette perspective ne manque pas d'inquiéter, sachant l'inadéquation avérée des activités de cette instance précédente.
La fonction de coordination interministérielle est certainement celle dont la nécessité et donc la justification sont les moins discutables. Il reste à s'assurer que la composition et le mode de fonctionnement interne permettront d'atteindre une certaine efficacité.
« L'analyse des événements susceptibles d'affecter la santé de la population » relève aussi de la compétence de l'InVS (cela a même été lourdement confirmé par la même loi), de la DGS au ministère et le cas échéant, selon les événements, des agences sanitaires. Les « doublonnages » sont donc prévisibles même si l'on prétend se placer à des échelons différents. Enfin, parfait mélange des genres, ce Comité national de santé publique est amené à contribuer à l'élaboration de la politique du Gouvernement dans les domaines de la sécurité sanitaire et de la prévention d'en examiner les conditions de financement , alors que l'on créé dans le même texte un Haut Conseil de santé publique . Donner des compétences d'orientation (pour le moins) en matière budgétaire à un tel organe consultatif est déjà inédit ; en outre, cela risque dès le départ de lui conférer une efficacité réduite par un positionnement institutionnel et administratif bancal.
Les réformes les plus récentes accroissent donc une compléxité publiquement dénoncée. On aurait pu espérer qu'elles donneraient un tout autre résultat. Cette remarque peut s'appliquer de la même façon à la création du département de gestion des situations d'urgences sanitaires de la DGS en son sein.
2.2.2. Le département des situations d'urgences sanitaires à la DGS
Lors de la réorganisation de l'été 2000 a été créé à la DGS un « bureau des alertes et des problèmes émergents ». Cette initiative a permis d'identifier un partenaire unique pour les partenaires extérieurs y compris l'InVS, mais il est vite apparu légitime aux services déconcentrés de le consulter devant toute situation anormale, ou de crise avérée, ce qui a favorisé les remontées d'information et les demandes de conseils. L'implication du bureau dans la gestion des situations exceptionnelles et les retours d'expérience qu'il a organisés ont conduit naturellement à lui confier l'élaboration des plans de réponse à des risques divers, dont les risques bioterroristes. L'élaboration de ces plans (variole, SRAS, grippe) représente une charge de travail considérable car il est nécessaire d'examiner une multitude d'éventualités et d'apporter des réponses concrètes (et plus seulement théoriques) à un niveau de détail très fin.
Ce bureau n'avait pas été dimensionné pour ce type d'activité. Le fait que son intitulé évoque « les problèmes émergents » montre bien que l'intention initiale était de développer une activité de prospective plus que de gestion opérationnelle des situations d'urgence. Or ce bureau s'est vite trouvé débordé au point de ne plus pouvoir faire face à la situation qu'il a en partie engendrée par son dynamisme.
C'est à la suite de ce constat que le ministre de la santé a annoncé en mars 2004 la création du « département des situations d'urgence sanitaires ».
Les éléments d'information recueillis à ce jour permettent d'apporter les précisions suivantes sachant que ce département « construit autour du bureau 5B préexistant aura pour perspective de devenir une sous-direction » et que l'effectif de départ de 20 personnes devrait être porté à terme à quarante.
On distingue ainsi trois missions : l'alerte, la préparation et les partenariats opérationnels.
-- la mission d'alerte
La première priorité est de consolider l'acquis du bureau 5B concernant le recueil et le traitement des informations et de travailler en lien étroit avec l'InVS à qui incombe l'analyse des informations et leur qualification en signaux d'alerte, ainsi qu'avec les services déconcentrés (ou les acteurs de santé) qui doivent gérer les situations.
Pour autant, ce nouveau département ministériel ne doit en aucun cas se substituer à la mission de l'InVS dont la loi relative à la politique de santé publique prévoit d'ailleurs le renforcement. C'est bien l'institut qui est en charge des systèmes de surveillance épidémiologique dont certains s'intègrent dans un cadre international. Cependant, toutes les alertes ne sont pas issues de tels systèmes. Les DDASS, les DSV, les DDCCRF, le COGIC (comité de gestion interministériel des crises), les autres agences comme l'AFSSAPS et l'AFSSA, les médecins, les établissements, les associations, les médias peuvent signaler des événements qu'il faut immédiatement traiter. Et c'est vers le ministère que ces acteurs se tourneront spontanément. Il y a donc besoin d'une équipe pour les traiter, les interpréter, alerter le ministre si nécessaire.
Il est indispensable que la DHOS, la DGAS et la DGSNR qui ont également créé des cellules de gestion des situations d'urgence soient étroitement associées à l'exécution de cette mission.
-- la mission de préparation
Elle consiste à développer les outils opérationnels permettant de mobiliser la société pour faire face à des situations d'urgences sanitaires. Cela concerne le système de santé, mais aussi la dimension sanitaire d'autres systèmes (transports collectifs, logements, écoles, etc ...).
En pratique, il s'agit essentiellement de l'élaboration et de la mise à jour des procédures correspondant au traitement de situations ponctuelles connues, d'une part, et des plans de réponse à des menaces plus générales (plan canicule, plan SRAS, plan grippe, plans répondant à des menaces terroristes ...), d'autre part. Cette activité devra être complétée par la conception d'exercices permettant de tester la faisabilité de certaines options et le caractère opérationnel des plans.
-- la mission des partenariats opérationnels et de la communication couvre en fait la structuration des liens pour la circulation des informations et le partage des tâches avant que l'urgence survienne et selon la typologie des urgences réalisée dans la mission de préparation.
La mission d'évaluation des quatre inspections dans son rapport précité (page 26) faisait part de ses doutes relatifs au positionnement de ce département vis-à-vis de l'InVS et vis-à-vis du COGIC :
-- L' « InVS, dont la mission, encore réaffirmée et précisée dans le projet de loi de santé publique, est de recueillir et d'interpréter tous les signaux d'alerte émanant soit des dispositifs de surveillance, soit des professionnels de santé. Or, dans la mesure où le nouveau département de la DGS aurait notamment pour mission « de recueillir et traiter les signaux d'alerte sanitaire, quelles que soient leur origine », on peut craindre un risque de doublonnage, voire de confusion, qu'il conviendra de réduire au maximum.
-- Le Comité de gestion interministérielle des crises (COGIC) de la direction de la défense et de la sécurité civiles qui est le lieu de veille permanente et de mobilisation des différents moyens de secours et d'intervention, dès lorsqu'une crise, sanitaire ou autre, nécessite une organisation de sa gestion au plan national. Le rôle de la sécurité civile, et la coordination des différents plans d'urgence selon un modèle uniformisé, sont d'ailleurs renforcés et réaffirmés par le projet de loi de modernisation de la sécurité civile, déposé récemment au Parlement. S'il est légitime et nécessaire que le ministère de la santé élabore les plans d'urgence destinés à faire face aux crises sanitaires ou aux composantes sanitaires des crises, et qu'il s'organise de manière à être en mesure de se mobiliser pour gérer la composante sanitaire d'une crise ou une crise à dominante sanitaire, la juxtaposition pure et simple au COGIC d'une structure de veille permanente propre au ministère de la santé est de nature à entraîner une duplication coûteuse de moyens ».
Il est à noter que la plupart des directions concernées par ce projet n'ont pas été appelées à une concertation par la DGS avant l'annonce de sa réalisation. Compte tenu des risques qui viennent d'être évoqués, cela aurait peut-être pu être évité.
2.2.3. Les CIRE
Les cellules interrégionales d'épidémiologie (CIRE) constituent le « bras » de l'InVS sur le terrain à la fois pour l'alerte, le suivi et la coordination de la veille au niveau régional. Elles sont naturellement appelées à jouer un rôle accru dans le cadre de la loi de santé publique (9 août 2004) avec les plans régionaux de santé publique et en particulier le plan d'action relatif à l'alerte et à la gestion des situations d'urgence sanitaire. La montée en puissance de ces structures, dont l'intérêt a été prouvé lors de l'épisode de légionellose dans la région de Lens en octobre-décembre 2003, se fait progressivement. 16 CIRE existent actuellement et emploient 71 scientifiques sur les nouvelles missions des CIRE ; le rapport annuel de l'InVS publié en octobre 2004 indique (page 53) :
« La mission fondamentale des CIRE évolue : le simple appui technique aux services déconcentrés de l'Etat qui leur était dévolu doit laisser la place à une véritable déclinaison des missions nationales de veille et d'alerte sanitaires au niveau régional. La place institutionnelle des CIRE doit parallèlement évoluer : il s'agit d'organiser la présence de l'InVS en région.
Dans ce cadre, pour le compte du GRSP, la CIRE sera le pilote du plan régional relatif à l'alerte et à la gestion des situations d'urgence sanitaire. Par ailleurs, la CIRE participera à l'élaboration du PRSP et aux travaux du GRSP. Au sein de ce Groupement d'intérêt public (GIP), l'InVS s'investira dans le diagnostic régional, l'identification des problèmes de santé régionaux et des indicateurs permettant le suivi des programmes, au travers de chaque CIRE qui apportera une référence méthodologique et scientifique à ces travaux, ainsi que sa compétence et son savoir-faire ».
La conception d'origine avancée pour les CIRE (« simple appui technique aux services déconcentrés de l'Etat ») laisse quelque peu perplexe car elle apparaît minorante. On veut espérer que leur rôle spécifique, leur autorité scientifique et à travers elles, celle de l'InVS, seront reconnus au niveau qui doit être le leur.
C'est aussi ce que souligne le rapport d'évaluation des quatre inspections (page 68) :
« Dans la perspective, d'une part de la participation des cellules interrégionales d'épidémiologie aux travaux du groupement régional de santé publique créé par la loi relative à la santé publique, groupement d'intérêt public présidé par le représentant de l'Etat dans la région et dont l'InVS est membre, d'autre part de leur inévitable implication dans l'élaboration du plan d'action relatif à l'alerte et à la gestion des situations d'urgence sanitaire, composante rendu obligatoire par l'article 10 bis de la loi précitée, du plan régional de santé publique arrêté par le représentant de l'Etat dans la région, il apparaît à la mission indispensable : d'asseoir les relations de l'Institut, des CIRE, des DRASS et des DDASS sur un protocole précisant les méthodes de travail et la répartition des tâches entre les différents acteurs ; de préserver l'autorité scientifique de l'Institut de veille sanitaire sur l'ensemble des travaux de la CIRE concernant la veille sanitaire et l'alerte, y compris le plan régional d'action ; de reconnaître à la CIRE un pouvoir fonctionnel d'animation et de formation des DDASS dans le cadre de leurs contributions à la fonction de veille et d'alerte ».
Cette mise en garde répond à une nécessité car le risque a été ou est réel d'une marginalisation fonctionnelle par rapport aux directions régionales et départementales de services du ministère est réel.
Enfin, les moyens des CIRE doivent être concentrés de la manière la plus efficace en évitant l'éparpillement tant que la masse critique d'efficacité n'a pas été atteinte pour une région ; il apparaît que les options prises par le directeur général vont dans le bon sens.
III. UNE CLARIFICATION NÉCESSAIRE
La problématique de la sécurité sanitaire dans ses multiples facettes a été bien appréhendée, que ce soit lors des travaux préparatoires de la loi de 1998, par les dispositions de la loi elle-même et pour les grandes orientations de sa mise en oeuvre. En revanche, les faiblesses non négligeables qui ont été observées montrent que certains éléments de l'architecture institutionnelle reliant les différents acteurs dans la veille et l'action ont besoin d'être révisés, voire modifiés. C'est ce qui est proposé dans le développement suivant et conclusif (IV - L'exhaustivité sans l'éparpillement). Au préalable, il convient ici de résumer les quelques principes nécessaires pour conférer à l'action publique une meilleure efficacité. Ils correspondent pour l'essentiel à une clarification méthodologique dont certaines crises ont montré l'urgence.
3.1. La distinction fondamentale : veille sanitaire/épidémiologie
La crise de la canicule a illustré de manière dramatique (cf. supra) les conséquences de l'oubli de cette règle de base. Au-delà des responsabilités des différents acteurs quant à l'inadéquation de certains mécanismes et le cloisonnement étonnant des structures administratives (au sein du ministère de la santé, avec l'administration préfectorale, notamment à Paris etc ...), c'est l'insuffisance de cette distinction qui doit être mise en cause. Un exemple particulièrement éclairant, a été fourni par la transmission des certifications de décès. Conçue dans une perspective d'analyse épidémiologique, la procédure de transmission avec les délais quelle comportait pour une formalisation complète (plus d'un an) ne pouvait évidemment en rien contribuer à quelque alerte ou même veille que ce soit. C'est encore un problème qui n'avait pas été signalé par les nombreux experts consultés dans le cadre de l'élaboration du plan national de sécurité sanitaire. Cette lacune est d'autant plus étonnante que la canicule avec l'hyperthermie maligne n'est pas le seul fléau pour lequel des données opérationnelles de décès en temps réel sont nécessaires. La mission d'évaluation des quatre inspections (op. cit. page 57) a fait de son côté la même observation dans les termes suivants :
« La mission a pu constater à maintes reprises, à l'occasion de ses investigations et des nombreuses auditions auxquelles elle a procédées, combien l'absence de cadre conceptuel partagé était source de malentendus et de désaccords entre les acteurs. Des notions aussi fondamentales que celles de surveillance, d'alerte, de mesure et d'évaluation du risque, sont susceptibles de recevoir une acception ou une portée différentes selon les interlocuteurs en présence, les textes en vigueur, la spécificité des domaines concernés, ou la traduction que l'on retient des concepts utilisés au niveau international ».
Le prix bien lourd de la crise de 2003 a été nécessaire pour découvrir cette exigence. On a tardivement retenu la leçon dès septembre 2003 puis dans la loi de santé publique elle-même. L'InVS lui-même semble avoir tiré des conclusions en ce sens ; cela apparaît clairement à travers son rapport annuel 2003 paru en octobre 2004, tout d'abord dans l'introduction du chapitre « Evolution des fonctions de veille et d'alerte de l'InVS à la suite des événements de l'année 2003 » (page 43) :
« L'année 2003 a connu beaucoup d'alertes sanitaires qui ont mis en tension les capacités de réponse de l'InVS. Le déroulement des événements et la manière dont ils ont été traités par la structure ont permis la réalisation d'analyses scientifiques et la rédaction de propositions utilisables pour la mise en oeuvre de politiques de santé.
La démarche suivie a conduit l'InVS aux conclusions suivantes :
-- premièrement, de nombreux événements qui se déroulent dans le monde peuvent avoir un retentissement sur la santé des habitants de la France (cette remarque paraît évidente pour les maladies infectieuses - Sras, grippe - mais elle concerne également d'autres phénomènes, notamment environnementaux). Il est donc indispensable de développer une veille internationale.
Parallèlement, les échanges rapides réalisés sur la planète, l'évolution climatique, l'augmentation de la pollution, les changements d'alimentation, le vieillissement de la population amènent à une évolution sensible de la santé humaine. Il nous faut anticiper la survenue de ces phénomènes que nous appelons « émergents ». Pour la prévision de phénomènes sanitaires à plus long terme, un regard large et multi-disciplinaire mettant en lien les sciences médicales avec les sciences sociales et de nombreuses autres disciplines permettrait d'appréhender l'évolution de notre environnement et son implication sur la santé humaine.
La « veille prospective » ainsi définie pourrait être mise en oeuvre dans les années à venir. Veille internationale, veille sur les phénomènes émergents et veille prospective aboutiront à la définition des priorités de travail de l'InVS à moyen et long terme.
-- deuxièmement, et de façon plus rapidement opérationnelle, l'InVS doit améliorer sa réponse à la mission d'alerte et de participation à la gestion des situations d'urgence. Il l'a fait dès l'année 2003 en mettant en place un système de surveillance spécifique à partir des services d'urgences et des données de mortalité, et il assure une organisation régionale de cette fonction d'alerte au travers d'un renforcement des CIRE qui se poursuivra dans les années à venir ».
Sans entrer ici dans le détail des mécanismes nouveaux mis en oeuvre ou en état d'être finalisés 45 ( * ) par l'InVS, celui-ci présente « l'intérêt d'une veille scientifique sur les phénomènes émergents prévisibles et d'une réelle prospective pluridisciplinaire sur les risques non identifiés ».
L'articulation nécessaire, mais aussi la distinction stricte entre la veille sanitaire et l'épidémiologie apparaissent ainsi clairement. Cela est d'autant plus important que l'acteur essentiel du système de sécurité sanitaire qu'est l'inVS a naturellement en charge à travers des unités différentes, des fonctions d'épidémiologiste (directes ou indirectes) et des fonctions de veille et d'alerte ; il n'est d'ailleurs pas seul dans ce cas.
C'est là une problématique que l'on est amené à retrouver par exemple dans la sphère de la santé du travail. En outre, la distinction doit s'imposer dans le cadre de l'affectation des moyens budgétaires car si le domaine de la veille sanitaire peut faire l'objet d'une délimitation relativement aisée une fois établi un système efficace, il n'en va pas de même avec l'épidémiologie pour laquelle des choix devront toujours faire l'objet d'arbitrages forcément difficiles.
3.2. La détermination des objectifs
* Les moyens mis à disposition des différentes instances doivent l'être d'une manière claire, rationnelle dans une perspective opérationnelle ; l'outil d'un tel ordonnancement est le COM (contrat d'objectifs et de moyens). On a noté la nécessité de son élaboration pour l'AFSSA et l'AFSSAPS (cf. supra) ; on a remarqué aussi qu'il ne saurait résoudre toutes les difficultés, mais qu'il reste en quelque sorte une « condition préalable » au fonctionnement de chacune des unités. Certaines, et notamment l'AFSSA, sont directement concernées par ce que l'on peut appeler « les ratés de l'interministérialité ». La partie « objectifs » du COM est essentielle dans la clarification de la démarche de procédure sanitaire qu'il s'agisse de l'action de chaque intervenant ou de la démarche d'ensemble.
* Cette détermination concertée des objectifs appelle une amélioration de l'exercice de la tutelle et des ministères qui, n'ayant pas juridiquement un pouvoir de tutelle, sont néanmoins des parties prenantes majeures dans la veille sanitaire, et ensuite dans l'action elle-même.
L'appareil d'analyse des situations concrètes et de quantification de l'action administrative doit être développé dans le cadre de ce COM. Les « capteurs » dont l'absence ou le non-fonctionnement ont été soulignés par le ministre de la santé en août 2003 se révèlent en effet indispensables. La démarche qui impliquera la LOLF (loi organique relative aux lois de finances) pourrait contribuer à de tels progrès. Cela posé, elle ne règlera pas les conflits récurrents de la conception et de la gestion interministérielle de certains domaines.
3.3. Un pragmatisme rationalisé
Les concepts étant clarifiés et les objectifs définis, les perspectives de retouches ou de modifications de l'architecture institutionnelle peuvent logiquement s'envisager sous réserve qu'en dernier lieu quelques règles simples, pour ne pas dire des recettes, restent présentes à l'esprit ; le terme de « pragmatisme rationalisé », aux antipodes d'un esprit de système théorique et complexe peut constituer le fil directeur de la démarche.
* Les résultats seuls constituent l'échelle d'évaluation d'une organisation et d'un jeu de mécanismes. Cette constatation peut paraître banale, mais doit être rappelée lorsque la qualité de l'organigramme et la complétude des procédures, tout à fait nécessaires, semblent le faire oublier. L'activité de l'AFSSA et de l'AFSSAPS, l'essentiel de celle de l'InVS (SRAS, légionellose, biotox notamment) en fournissent une illustration positive.
* La constatation d'un besoin de réforme ne saurait justifier des modifications de structures faites au coup par coup. L'expérience montre d'ailleurs que pour corriger un dysfonctionnement, on a parfois tendance à commettre une première erreur, puis pour corriger celle-ci une seconde. Ce qui s'est passé avec le FOPIM, avec les transferts de la commission de la Transparence dans le domaine du médicament, avec le CNSS et l'ensemble des organes consultatifs dans le domaine de la santé publique est éclairant.
* La complexité doit donc être évitée car elle peut être fatale. Cette remarque peut d'ailleurs s'appliquer utilement à des procédures européennes où les risques de contradictions internes et/ou communautaires sont réels (domaine alimentaire notamment) et où l'efficacité des contrôles et de leur coordination est problématique. Or, il s'agit là d'éléments essentiels de la sécurité sanitaire. On peut tout à fait admettre que des opérations d'expertise exigent pour être efficaces et sûres des procédures subtiles ; en revanche lorsque la complexité n'est pas due à la nature même du problème posé, elle est d'autant moins acceptable qu'elle devient nécessairement source de risque.
* La faisabilité des modifications institutionnelles implique aussi de la part des autorités ministérielles et d'une manière générale du Gouvernement, un réalisme dans les projets et les exigences.
La fixation de calendriers irréalistes pour satisfaire des « effets d'affiche », même si cela n'est certes par nouveau, reste à écarter.
Les moyens, budgétaires notamment, ne sauraient être considérés comme secondaires ; cela peut d'ailleurs s'apprécier de différentes manières ; on pense naturellement à l'insuffisance, mais la surestimation de la capacité d'organisation ou l'exigence d'un calendrier excessif peuvent amener une situation d'excès de crédits : la mise en place de l'AFSSAPS en fournit un exemple. L'appréciation des moyens humains exige aussi que la faisabilité soit bien cernée. Dans certains domaines et/où certaines fonctions, il a été noté que l'insuffisance du vivier d'experts pouvait constituer un frein, voire une menace, comme dans le cas de la toxicologie. Le problème est là très difficile car la situation exige d'agir avec obligation de résultats, sans que l'on puisse intervenir sur une partie du mécanisme où les instances administratives n'ont pas prise. La dimension essentielle du problème n'est d'ailleurs pas ici d'ordre budgétaire, mais méthodologique.
Enfin, l'appréciation des moyens doit nécessairement se faire en rapport avec la perspective des plans de charge. De ce point de vue, une création comme celle de l'AFSSE montre clairement que ces quelques recettes de « pragmatisme rationalisé » ont une valeur permanente.
IV. L'EXHAUSTIVITÉ SANS L'ÉPARPILLEMENT
4.1. Les lacunes
Il peut paraître surprenant qu'après la création des agences de sécurité sanitaire mises en oeuvre depuis plus de six ans, des lacunes ou des « zones blanches » puissent encore exister. Le paradoxe apparent s'explique par des facteurs très divers.
4.1.1. Les produits chimiques
On trouve tout d'abord des produits dont l'analyse n'a été ni organisée ni même prévue dans une perspective de risque général.
* C'est le cas des substances et produits chimiques dont le nombre, enregistré au niveau européen s'élève à près de 100.000. Ces produits interviennent naturellement dans les risques sanitaires d'origine professionnelle. Toutefois, leur analyse ne devrait pas se limiter à cette seule perspective. Des événements surviennent fréquemment pour rappeler que le milieu de vie quotidienne est aussi confronté à ces risques ; d'ailleurs la distinction entre les deux sphères est de plus en plus dépourvue de consistance. La même substance peut être utilisée en usine, à la maison par un bricoleur ou par des personnels chargés de l'entretien ou du nettoyage etc ... Le recours incontrôlé à l'amiante a indiqué il y a déjà longtemps que beaucoup de risques ne se segmentent pas.
La manière dont l'Union européenne tente d'appréhender le problème en illustre d'ailleurs la difficulté. Celle-ci a opéré une distinction entre substances anciennes et substances nouvelles, ces dernières étant systématiquement notifiées auprès d'une autorité nationale en France, le ministère de l'écologie et l'INRS). Depuis 1981, 5000 substances nouvelles ont ainsi été notifiées et examinées.
Pour les secondes, que la Commission évalue à 30.000, leur évolution devrait se faire progressivement dans le cadre d'un règlement communautaire Reach 46 ( * ) . Son adoption est prévue pour le courant de l'année 2006. Sa mise en oeuvre nécessitera au moins une huitaine d'années. La perception de ces difficultés est d'ailleurs grande en Europe, ainsi que cela a pu être constaté sur le terrain.
* Des équipements, dispositifs, matériels utilisés dans la vie quotidienne dans des situations très diverses peuvent comporter des risques dont l'appréciation reste mal définie. La compétence générale de la DGCCRF (ministère chargé de la consommation) existe et, depuis décembre 2002, l'AFSEE peut être saisie sur nombre de ces questions ; cette dernière a d'ailleurs été saisie sur les téléphones portables à deux reprises (cf. supra). Sur le danger des cabines de bronzage, la DGCCRF est amenée à vérifier la mise en oeuvre de la réglementation existante, mais le cadre même dans lequel le problème est posé ne correspond pas aux exigences de rigueur et de sécurité : ici c'est le principe même de l'analyse, des critères et l'ensemble de la procédure ou plutôt de l'absence de procédure qui sont en cause.
4.1.2. Les produits phytosanitaires
Les produits phytosanitaires constituent une catégorie couverte par des procédures existantes, complexes et très parcellisées, mais qui sont restées en dehors du périmètre que la loi de 1998 a déterminé pour l'AFSSA. La procédure d'évaluation du risque reste dans le domaine de la DGAL du ministère de l'agriculture qui a également en charge le secteur économique. Ici la crise n'est pas due aux textes, mais à leur non respect (cf. supra sur l'AFSSA).
4.1.3. La sécurité sanitaire au travail
La sécurité sanitaire en milieu de travail apparaît aussi comme une zone où les risques peuvent être insuffisamment, voire mal cernés et donc mal traités. La direction des relations du travail du ministère du travail est en charge du suivi de ce domaine et peut s'appuyer principalement sur l'InVS et l'INRS. Mais elle le fait dans des conditions qui sont critiquées par le rapport d'évaluation des quatre inspections (op. cit. page 29) :
« L'Institut de veille sanitaire en matière de veille et de surveillance, au travers de son département « santé au travail », mais également de son département « santé-environnement ». Le département « santé au travail » de l'InVS est le plus petit département de l'InVS (16 personnes, soit 5 % environ des effectifs totaux). La Direction des relations du travail n'exerce qu'une magistrature d'influence vis-à-vis des activités de ce département, puisqu'elle n'a pas la co-tutelle de l'Institut et ne contribue à son financement que de façon marginale (à hauteur de 120K€ par an environ, pour le programme de surveillance du mésothéliome). Elle estime en particulier que l'activité de l'institut reste trop axée sur des études épidémiologiques à visée universitaire, déterminées de façon autonome et pas assez sur les outils structurants de surveillance de la santé de la population, dans une perspective de veille et d'alerte ».
« L'Institut national de recherche et de sécurité pour la prévention des accidents du travail et des maladies professionnelles (INRS), structure associative aux moyens importants (700 personnes, 80 M€ de budget), mais placée sous l'égide de la caisse nationale d'assurance maladie des travailleurs salariés (CNAMTS) et gérée par un conseil d'administration paritaire associant des représentants des employeurs et des syndicats de salariés (son financement provenant presque exclusivement du fonds national de prévention des accidents du travail et des maladies professionnelles). Même si les relations de cet organisme sont dans l'ensemble bonnes et suivies avec la direction des relations du travail, cette dernière ne disposant ni des moyens d'intervention d'une autorité de tutelle ni de levier de financement significatif, n'est pas vraiment en mesure d'influencer fortement les priorités et les programmes de travail de l'INRS, dès lors que les actions à mener sortent du champ de compétence normal de l'assurance maladie et relèvent d'une responsabilité d'Etat ».
* L'urgence qu'il y a à résoudre cette question de la sécurité sanitaire en milieu professionnel n'est pas nouvelle, mais elle est devenue d'autant plus forte qu'elle a été récemment soulignée par les décisions rendues le 3 mars 2004 par le Conseil d'Etat statuant en cassation sur la responsabilité de l'Etat dans l'affaire de l'amiante.
La Cour administrative d'appel, puis le Conseil d'Etat, estiment que les autorités publiques n'avaient pas entrepris suffisamment tôt de « recherche afin d'évaluer les risques pesant sur les travailleurs exposés aux poussières d'amiante » et pas davantage « pris de mesures aptes à limiter ou éliminer les dangers ». La responsabilité de l'Etat a été ainsi clairement identifiée. La nécessité de disposer d'une structure d'analyse, d'évaluation des risques dans ce domaine s'impose donc, au besoin en changeant explicitement des instances existantes et en mettant à leur disposition les moyens juridiques et matériels correspondants.
4.2. Les tentations
Face au foisonnement de difficultés de mise en oeuvre, de lacunes et de chevauchement de compétences, plusieurs tentations apparaissent. Elles peuvent correspondre à la résurgence d'idées anciennes ou aux enseignements hâtifs de crises récentes.
4.2.1. La tentation unificatrice
* Une première tentation est celle de la création d'une agence de sécurité sanitaire unique. Cette hypothèse avait été sérieusement envisagée en 1997-1998, notamment par M. Bernard Kouchner alors ministre de la Santé (cf. supra 1 ème partie). L'expérience a plutôt validé une organisation différente et il ne paraît pas actuellement envisageable de revenir sur ce choix. Une FDA à la française est moins que jamais séduisante, même si l'existence de plusieurs agences met particulièrement en lumière les difficultés de l'interministérialité. Ce dernier argument est utilisé à l'appui d'une seconde voie.
* Cette deuxième tentation vise à unifier l'autorité ministérielle. Il s'agit là du schéma avancé par l'ancien directeur de la DGS, le Pr. Lucien Abenhaïm dans son livre « Canicules » plus précisément dans son chapitre 10 (« la sécurité sanitaire inachevée »).
Certains aspects de ce schéma ont déjà été abordés dans le présent rapport (cf. 1 ère partie). La proposition centrale consiste en la création d'un « ministère de l'Environnement et de la sécurité sanitaire » qu'il évoque ensuite par l'expression « grand ministère des risques ». Il précise ainsi cette proposition (p. 248 et 249)
« Il faudrait notamment, pour éviter tout conflit de compétences, regrouper l'environnement, l'alimentation, la consommation et les produits, sans oublier les questions de la santé au travail. Un ministère regroupant l'ensemble des composantes de la sécurité sanitaire serait donc considérablement plus efficace. Il devrait reposer sur des agences indépendantes chargées de l'évaluation et de la gestion des risques. Pour parvenir à ce résultat, il faudrait tout d'abord que l'Agriculture se départisse de la sécurité alimentaire, les Finances de la consommation et la Santé de la gestion des risques des milieux, ainsi en fait que de toute la sécurité sanitaire des produits. Tous ces secteurs seraient ensuite regroupés au sein de notre grand ministère - rejoints par la sûreté nucléaire et la radioprotection - et répartis entre les agences ».
M. Abenhaïm supprime le principe de séparation entre les fonctions de gestion et d'évaluation du risque (quelle que soit la dénomination exacte que l'on retienne pour cette dernière fonction qui peut en effet varier) et, propose la création d'un « grand ministère » qui regrouperait de nombreux domaines. Celui-ci perdrait en fait l'essentiel de ses compétences au profit « d'agences indépendantes » et non « autonomes ». « L'indépendance » de ces agences ne serait pas un vain mot puisque l'auteur de ce schéma propose une méthode de désignation de leurs dirigeants hors du champ ministériel :
« Voilà pourquoi je pense que les directions des agences devraient être sélectionnées par leurs conseils d'administration et non par des ministères, après des appels à candidature publics et en s'appuyant sur des comités également indépendants ou siègeraient notamment des scientifiques. Leur position et leur autorité en seraient renforcées ».
Cette perspective appelle naturellement quelques questions. Comment les membres des conseils d'administration seraient-ils désignés ? S'il s'agit de représentants des ministères, il ne s'agit que d'un habillage différent qui peut difficilement être une marque de progrès et un gage d'efficacité. S'il ne s'agit pas de cela, quelle serait alors la légitimité des membres de ces conseils ? Peut-on vraiment envisager de confier l'exercice de responsabilités d'autorités publiques à des « comités indépendants » ? , indépendants de qui ? Des domaines politiques dont la légitimité démocratique est établie, mais peut-être pas d'écoles scientifiques, de chapelles de divers ordres, d'associations en tout genre, d'intérêts économiques et sociaux ouverts ou dissimulés ...
Derrière un débat en apparence technique se profile une question sur la conception de l'Etat, sur ses responsabilités inaliénables, et sur une certaine approche la République. Au-delà d'une mode qui abandonne volontiers les prérogatives naturelles de l'Etat, on ne perçoit pas dans cette approche une réponse à la recherche d'efficacité en matière sanitaire.
4.2.2. La tentation de parcellisation
La persistance et l'apparition de difficultés, à l'occasion de « crises sanitaires » notamment, sont à l'origine de réponses institutionnelles qui se sont traduites par la création de nouvelles instances souvent formalisées en agences. On pourrait caricaturer ce qui deviendrait en quelque sorte un réflexe : un problème, une agence : agence de biomédecine, agence de santé des végétaux, de la santé au travail. Cette dérive correspond non seulement à une mode, mais encore à une sorte de renoncement réel ou apparent. Dans d'autres cas encore, il s'agirait d'autonomiser une fonction, inutilement voire dangereusement, pour satisfaire un « effet d'affiche » . Dans d'autres situations encore, une « nouvelle agence » peut constituer un moyen tactique de diversion dans les combats de l'interministériel.
L'AFSSAPS constitue d'ailleurs une bonne illustration de la capacité à regrouper au sein d'une entité logique des compétences dont certaines auraient pu faire l'objet d'une agence séparément.
Pour toutes ces raisons, la multiplication des agences est à éviter, d'autant que des regroupements opérationnels, peuvent être envisagés sous d'autres conditions.
4.3. Les solutions envisageables
Les structures et les périmètres de compétences des trois principales entités créées par la loi de 1998, l'InVS, l'AFSSA et l'AFSSAPS, constituent des repères validés par l'expérience. La majorité des observateurs s'accorde à les trouver adaptés aux objectifs initiaux et, pour l'essentiel, aux besoins constatés aujourd'hui. On a vu que certaines corrections restent à faire avec l'exercice effectif des compétences de l'AFSSE.
4.3.1. L'AFSSE et ses domaines
La loi du 9 mai 2001 avec la création de l'AFSSE visait à résoudre ces problèmes qui avaient déjà été identifiés. Elle n'a pas, d'évidence, pu constituer une solution, ni même la piste d'une solution.
Tout d'abord, la mise en place de l'AFSSE s'est réalisée dans des conditions qui ne lui donnaient pas tous les atouts au départ :
? installation officielle en septembre 2002, les financements minimum n'étant acquis que trois mois plus tard ;
? corrélativement des moyens humains très limités (une trentaine de personnes à ce jour)
? et une installation matérielle dans un provisoire qui semble appelé à durer.
En fait, l'Agence est en état de fonctionner depuis à peine deux ans. Ces considérations ne sauraient évidemment être retenues à son débit, d'autant qu'elle s'est immédiatement mise au travail en constituant ses comités d'experts spécialisés et en traitant les premières saisines. En outre, et cela a été une lourde tâche, l'AFSSE a été en charge de l'élaboration du PNSE (Plan national santé environnement) achevé au début de l'année 2004 et qui constitue désormais une base de réflexion et d'action.
Ce très faible recul ne facilite donc pas l'évaluation de l'application de la loi de 2001. C'est la raison pour laquelle il n'y a pas été procédé ici d'une manière systématique. En revanche, l'appréciation des difficultés qui se révèlent dans l'ensemble des domaines relevant de la compétence de l'AFSSE montre que des solutions doivent être trouvées sans attendre le résultat d'une telle évaluation. Plus important encore, il apparaît clairement que le positionnement de l'AFSSE par la loi, dès le départ, n'a pas été le bon. Les intentions qui ont présidé à sa création procédaient de philosophies parfois différentes et cette création s'est donc engagée dans une certaine ambiguïté.
Pour prendre un seul exemple, des difficultés du positionnement de principe de l'AFSSE, la loi (art. L. 1335-3-1 du code de la santé publique) disposait qu'un décret en Conseil d'Etat devait « fixer la liste des établissements publics de l'Etat qui apportent leur concours permanent à l'agence », et que « dans un délai d'un an au plus tard après la publication de la loi, chacun de ces établissements négocie avec l'agence la mise à disposition de celle-ci de ses compétences et de ses moyens d'action ». Ce même décret devait « fixer également les modalités selon lesquelles l'agence coordonne et organise les missions d'évaluation conduites par les autres organismes intervenant dans son champ de compétence ».
Or cette mise en oeuvre n'a pas eu lieu ; certes, le décret du 1 er mars 2002 (art. R.795-2 du CSP) a bien déterminé 15 établissements publics supposés mettre à disposition de l'AFSSE compétences et moyens ; deux conventions ont été à ce jour passées avec l'INERIS et l'INSERM.
Le rapport d'évaluation des quatre inspections a ainsi développé le constat des difficultés de principe que pose l'interface AFSSE/InVS(page 82) et qui s'impose à tous les observateurs :
« Les rôles respectifs de l'AFSSE et de l'InVS, dont le département « santé-environnement » préexistant à la création de l'AFSSE développe une activité importante, aussi bien en matière d'identification ou de caractérisation de facteurs ou de situations à risques, qu'en matière d'évaluation de l'impact sanitaire des pollutions chroniques ou accidentelles ne sont toujours pas clarifiés. Les débats entre les deux organismes sont toujours à l'heure actuellement d'une grande vivacité. L'AFSSE a proposé une répartition des attributions, selon une « polarisation » qui ferait de l'InVS une « porte d'entrée » naturelle lorsque le point d'entrée est d'emblée sanitaire (suspicion ou allégation d'un cluster, signaux épidémiologiques issus de la surveillance par exemple), et de l'AFSSE l'opérateur de « première ligne » lorsqu'une question environnementale constitue le « point d'appel » (qualité du milieu, rejets industriels par exemple). L'InVS considère une telle démarcation à la fois « peu réaliste » et juridiquement difficile à tenir pour l'Institut. Peu réaliste, dans la mesure où, d'une part, « tout dossier où une population est exposée nécessite une réflexion sur les dangers des facteurs environnementaux, une évaluation des expositions, un recueil de données sur les effets sanitaires, voire la mise en place d'un système de surveillance », et où, d'autre part, l'InVS auquel la loi confie notamment la mission de :a) ;rassembler, analyser et actualiser les connaissances sur les risques sanitaires, leur cause et leur évolution ; b) détecter de manière prospective les facteurs de risque susceptibles de modifier ou d'altérer la santé de la population ou de certaines de ses composantes, de manière soudaine et diffuse ». En d'autres termes, l'InVS considère qu'en matière de santé environnementale, il n'est généralement pas possible de séparer surveillance, alerte et évaluation du risque (hormis l'évaluation a priori des substances et des produits), les fonctions de surveillance, de détection et d'alerte devant être fondées sur des hypothèses quant aux facteurs de risque et supposant la capacité d'analyser le risque pour valider la pertinence d'un signal d'alerte ».
Des schémas de développement de l'AFSSE visant à lui permettre d'atteindre les objectifs fixés ont été envisagés, notamment par sa directrice générale ; ils comprendraient par exemple l'intégration de moyens actuellement situés à l'INERIS, au BRGM et à l'InVS, voire d'autres établissements pour permettre une expertise en interne, au-delà des validations d'expertises réalisées dans d'autres structures. Ce genre de proposition rendrait encore plus complexe et pour tout dire inextricable des domaines qui ne le sont déjà que trop.
Les constats auxquels on parvient convergent vers le non-maintien de l'AFSSE. La mission d'évaluation des quatre inspections a d'ailleurs listé l'essentiel de ces constats dans des termes qui résument la question (ibidem page 85) :
« Mais la mission considère, au terme de son enquête, qu'aucun de ces arguments ne paraît à la mission de nature à remédier aux problèmes de fond que la création de l'AFSSE n'a pu régler :
- les difficultés à définir une ligne de partage de compétences cohérente avec l'InVS ;
- la complexification des circuits induite par le positionnement de l'AFSSE à la fois en tant qu'organisateur d'une expertise susceptible d'être rationalisée par d'autres voies (cf. recommandation suivante), et en tant que producteur d'expertise et de connaissances (la préparation du plan national santé environnement, qui a donné l'occasion à l'agence de tenir le secrétariat des travaux de la commission d'orientation, ne pouvant guère faire illusion) ;
- l'accentuation des problèmes de coopération et de coordination posés par la multiplication des interfaces entre organismes à champs de compétences frontaliers, interpénétrés ou concurrents ;
- la difficulté structurelle des autorités publiques à assurer efficacement leurs missions de supervision stratégique, de coordination et d'évaluation d'un dispositif à opérateurs multiples ;
- la dispersion des moyens : d'une part, la réunion au sein de l'AFSEE en vue de la doter d'une taille critique et d'une plus grande capacité d'expertise interne, des compétences en sécurité environnementale existant dans d'autres structures (InVS, INRS, INERIS ...) n'est guère envisageable pour des raisons à la fois techniques, tenant aux missions mêmes des organismes susceptibles d'être concernés, et juridiques (statut de l'INRS) ; d'autre part, il serait contre-productif, à supposer que ce soit financièrement possible, de dupliquer au sein de cette agence l'expertise par ailleurs disponible, en matière de produits, à l'INRS et à l'INERIS) ;
- la meilleure prise en compte nécessaire de la protection des écosystèmes qui n'est pas mieux garantie par l'existence d'une agence de sécurité sanitaire spécifique pour l'évaluation des risques d'origine environnementale, et qui mériterait une réflexion sur l'opportunité d'instituer un opérateur dont les missions porteraient sur la protection de l'environnement en général.
C'est pour cet ensemble de raisons que la mission estime que les difficultés de mise en place de l'AFSSE s'expliquent non par des facteurs d'ordre conjoncturel, mais bien par des défauts de conception structurels, et qu'elle considère que la tentation d'accorder à l'AFSSE un délai d'installation dans le dispositif supplémentaire ne constituerait pas une solution pertinente à long terme ».
Le pragmatisme qui a été préconisé précédemment (cf. supra) pour conforter sans les bouleverser des institutions et des mécanismes existants doit de la même manière inspirer les choix de la puissance publique, mais en faisant prévaloir une architecture institutionnelle viable, adaptée à des réalités évolutives.
4.3.2. Une architecture simplifiée
Dans le droit fil des analyses précédentes, il apparaît que le dispositif global des agences, instances et mécanismes n'a pas à être modifié dans son architecture.
* L'AFSSAPS, dans ses pouvoirs, ses compétences et son périmètre, ne connaîtrait pas de changement. Sur ce dernier point quelques légères retouches peuvent être cependant envisagées avec profit pour certains « produits frontières », notamment pour mettre fin à l'existence de certaines « zones blanches ».
Il ne paraît donc pas souhaitable de transférer à l'AFSSAPS l'Agence du médicament vétérinaire, pour des raisons déjà détaillées (cf. supra pages) et parce qu'un tel transfert paraît contraire à l'esprit de la nouvelle structuration.
* L'InVS ne serait pas profondément restructuré ; ses compétences confirmées et précisées par la loi de santé publique du 9 août 2004 seraient pérennisées ; toutefois, il connaîtrait des modifications destinées à garantir l'exercice effectif de certaines fonctions qui pourraient être acquises, conséquences de la disparition de l'AFSSE.
* L'AFSSA elle-même dont le périmètre, les compétences et pouvoirs ne seraient pas non plus modifiés dans la sphère des produits alimentaires (avec le médicament vétérinaire), se verrait confier la « sécurité sanitaire des milieux de vie ». Les compétences actuelles de l'AFSSE qui peuvent être couvertes par cette expression deviendraient ainsi un domaine identifié et spécifique de la nouvelle agence qui s'ajouterait à son domaine actuel.
Les raisons qui militent en faveur d'un tel regroupement sont fortes, mais les réserves qu'il peut susciter ne doivent pas être ignorées ou même minimisées.
-- Parmi les arguments favorables, on relève d'abord l'expérience acquise par l'AFSSA et plus spécialement la DERNS dans l'analyse et l'évaluation des risques. On ne peut que renvoyer à la présentation faite précédemment des travaux de l'Agence dans son « coeur de métier » et qui, de l'avis général, sont une réussite. Les problématiques auxquelles les organismes d'évaluation du risque sont confrontés dans le domaine alimentaire et dans celui des milieux de vie présentent de réelles analogies.
Un secteur illustre particulièrement l'adéquation d'un tel regroupement : celui des produits phytosanitaires. Dans le cadre de la législation actuelle (loi du 1 er juillet 1998), ces produits rentrent indiscutablement dans le champ de compétences de l'AFSSA. Au moment où le maintien d'un ordonnancement contraire à la loi devient intenable avec les arrêts successifs du Conseil d'Etat sur le Régent et le Gaucho à l'encontre des décisions du ministère de l'agriculture, ce dernier sort un projet inconnu « d'Agence de santé des végétaux » . L'argumentation en faveur d'une telle idée est particulièrement légère : on y évoque le fait que la santé des plantes ne peut pas être vue sous le seul angle alimentaire, qu'elle fait question par elle-même et que l'environnement dans son ensemble est intéressé. Dès lors, on se tourne vers l'AFSSE. Mais on devrait également se tourner vers une agence de la sécurité au travail si celle-ci, d'après certaines annonces du ministre compétent, venait à être créée ; et d'ailleurs se pose, quelles que soient les structures, le problème des effets des produits phytosanitaires sur ceux qui les manipulent directement, et ceux qui ensuite, dans l'environnement général, sont à leur contact. Le caractère inextricable de la situation est ici particulièrement parlant. Au moins trois agences sanitaires seraient compétentes, sans parler d'autres instances spécifiques avec lesquelles elles seraient amenées à travailler.
Pour rester dans ce domaine, l'évolution du dossier Régent et Gaucho, a amené logiquement les trois directions ministérielles concernées (DGAL, DGS, DGCCRF) à saisir conjointement l'AFSSA et l'AFSSE de l'ensemble du dossier. Le mouvement se prouvant en marchant, on a là une preuve significative de l'intérêt du regroupement proposé entre ces deux agences.
-- La clarification que permettrait un tel regroupement est appréciable à la lumière des constats de la présente évaluation et aussi à la lumière de certains projets réellement hasardeux de création de nouvelles agences.
-- Les réserves qu'un tel regroupement peut susciter sont de trois ordres : hétérogénéité, taille excessive, complexité de certains processus d'évaluation.
L'hétérogénéité des situations, risques et produits n'est pas niable : la pollution due aux naufrages de pétroliers, les risques que font courir les cabines de bronzage ou ceux que peuvent recéler les produits phytosanitaires, sans parler de l'ESB ou du semicarbazide n'ont pas grand-chose en commun. Pour autant, les méthodologies adaptées aux différents secteurs peuvent procéder en fonds commun. En outre, il s'agit là d'une hétérogénéité que l'on rencontre déjà au sein de l'AFSSAPS.
? La taille excessive de la nouvelle agence peut constituer un argument d'opposition et en tout cas susciter des doutes ; si la parcellisation est à bannir, le gigantisme l'est également. Aussi plusieurs précautions apparaissent-elles souhaitables.
D'abord, l'identification des domaines au sein de la nouvelle agence peut et doit être claire et articulée d'une manière opérationnelle. A cet égard, l'AFSSAPS récemment restructurée constitue sinon un modèle, du moins une référence de méthodes et de pratiques.
En second lieu, cette nouvelle architecture ne se conçoit qu'en liaison avec la création parallèle du Groupement d'Intérêt Public (GIP) « substances et produits chimiques » proposé par le rapport d'évaluation des quatre inspections générales. Ce pôle d'expertise sur les produits permettait seul de faire face aux besoins identifiés par le programme européen Reach et à l'insuffisance inquiétante des moyens de la toxicologie. Cette proposition particulièrement nécessaire est présentée intégralement ci-après ; elle paraît, en outre, de nature à éviter le problème du gigantisme que la nouvelle agence pourrait, à défaut, poser :
« Réunir les compétences des différents acteurs en matière d'évaluation des substances chimiques
La structuration des capacités en matière d'expertise sur les substances chimiques est le deuxième volet du renforcement de la capacité de gestion du risque sanitaire en milieu professionnel et dans les autres milieux de vie. Comme il a été exposé plus haut (cf. 1.2.3.), les capacités françaises sont aujourd'hui déjà insuffisantes pour faire face à ses obligations européennes, et le seront encore plus si le projet de règlement REACH voit le jour, ce qui est un scénario probable.
La faiblesse du dispositif français, rappelons-le, tient à deux causes, s'alimentant l'une l'autre : la relative dispersion des moyens entre l'INRS et l'INERIS principalement, mais aussi entre les différentes agences de sécurité sanitaire et d'autres structures (commission d'étude de la toxicité par exemple) qui ont besoin pour leur mission propre de compétences en toxicologie ; la faiblesse de la discipline toxicologique en France, constatée tant en écotoxicologie et en toxicologie expérimentale qu'en toxicologie clinique (ce dont témoigne d'ailleurs la situation en termes de ressources humaines des centres anti-poison et centres de toxico-vigilance, mise en lumière par e rapport conjointement présenté par l'AFSSE et l'InVS en septembre 2003).
Un rapprochement des deux centres d'expertise principaux constitués par l'INRS et l'INERIS, s'il pourrait apparaître souhaitable dans l'absolu, se heurte à la difficulté de la différence de statut des deux instituts, due à la particularité de celui de l'INRS, association dépendante du régime des accidents du travail et maladies professionnelles. Il n'est guère envisageable de requérir les partenaires sociaux, gestionnaires de l'INRS, d'assumer une mission relevant de l'Etat. Il n'est guère concevable non plus, dans la conjoncture présente, de mobiliser des financements d'Etat importants en substitution à e que le régime AT/MP consacre aujourd'hui à la prévention des risques engendrés notamment par l'usage des produits chimiques.
De plus doit être prise en compte l'importance qu'il y a à maintenir les synergies développées à l'INERIS en matière de toxicologie et d'écotoxicologie, nécessaires à l'atteinte des « objectifs associés de sécurité sanitaire et de protection de l'environnement ».
La solution intermédiaire préconisée par la mission est donc celle d'un regroupement des moyens en analyse toxicologique aujourd'hui dispersés entre différents acteurs, via la création d'un groupement d'intérêt public, seule solution juridique permettant de préserver la place (et les financements) de chacune des institutions et de construire une capacité d'expertise à la fois renforcée et organisée dans le domaine des risques liés aux substances et produits chimiques. Un tel groupement, en outre, serait de nature à concilier deux objectifs : rassembler et coordonner les diverses ressources d'expertise mobilisables de manière à atteindre un effet de masse critique ; préserver et valoriser la spécialisation des experts dans leur domaine de compétence. Cette structure devrait par conséquent être ouverte largement à tous les organismes et structures de recherche impliqués dans ce domaine : INSERM, INRA, laboratoires universitaires (Pharmacie, Sciences ...), laboratoire d'écoles vétérinaires ... En effet, au-delà de l'intérêt à atteindre l'effet de masse critique nécessaire à une structuration et à une meilleure organisation de l'expertise en France, ainsi qu'à une action plus marquée à l'échelle internationale, ce regroupement devrait également permettre de développer des recherches destinées notamment à faire évoluer l'approche toxicologique traditionnelle - substance par substance, produit par produit - au profit d'une approche plus globale, prenant en compte l'analyse des effets cumulés ou des interactions en jeu, l'ensemble des voies d'exposition et les modes opératoires.
Il conviendrait que cette structuration nationale s'appuie également sur une rénovation du réseau des centres anti-poison et des centres de toxico-vigilance, qui suppose sans doute un renforcement de leurs moyens humains. Cette évolution est indispensable pour assurer le développement des capacités d'expertise en toxicologie clinique et renforcer les réseaux de vigilance 47 ( * ) . Ce réseau des centres anti-poison et centres de toxico-vigilance devrait s'inscrire à l'avenir dans le réseau des centres de référence et réseaux sentinelles coordonnés par l'InVS, avec les mêmes modes financiers de conventionnement pour leurs missions d'expertise et d'alerte. »
? La complexité des processus d'évaluation, notamment lorsque trois structures sont amenées à intervenir (InVS, nouvelle agence, pôle d'expertise extérieur) n'est pas niable, mais elle serait inférieure à la situation actuelle surtout et elle pourrait être rationalisée, voire diminuée par l'élaboration de procédures rigoureuses entre des organismes aux compétences clairement déterminées, qu'il s'agisse du pôle d'expertise ou de l'InVS. Au sujet de ce dernier, cette délimitation des missions est effectivement l'une des conditions de la réussite du regroupement AFSSA-AFSSE en une nouvelle agence.
La nouvelle précision apportée aux missions générales de l'InVS par la loi de santé publique du 9 août 2004 (cf. supra) indique clairement le champ très large de ses compétences de veille proprement dite : surveillance et investigation des facteurs de risque et des expositions, enquête sur des risques spécifiques locaux ou sectoriels, et déclenchement des alertes. La nouvelle agence, chargée strictement de l'évaluation aurait pour mission l'expertise et l'évaluation des risques a priori (substances et produits nouveaux, des produits de consommation, risques sanitaires d'installations, d'équipement, de procédés, d'agents physiques).
Une dernière difficulté peut se profiler avec le périmètre de la nouvelle agence : celle des tutelles, de leur nombre et de leur condition d'exercice. On a noté au sujet de l'AFSSA les difficultés d'exercice de la tutelle de l'Etat du fait de la multiplicité des ministères. On peut imaginer les situations complexes que l'existence d'au moins six tutelles pourrait entraîner. Cette difficulté ne devrait pas constituer un obstacle dirimant, mais devrait être soigneusement analysée et résolue avant l'élaboration de l'architecture institutionnelle retenue.
La réalisation de ce diagnostic préalable à la décision amènera aussi les administrations ministérielles à préciser les fonctions qui restent dans leurs domaines respectifs et d'éviter à l'avenir des interférences entre évaluateurs et gestionnaires des risques et, a fortiori, opérateurs.
RÉSUMÉ DE LA QUATRIÈME PARTIE
|
La réforme de 1998, pour profonde et novatrice qu'elle soit, n'a pas apporté toutes les simplifications que l'on pouvait espérer. Elle a maintenu des organismes dont les compétences se recoupent, mais surtout elle a été suivie par des modifications qui ont souvent encore compliqué la situation. Une clarification s'avère nécessaire, d'une part, pour l'ensemble du champ de la sécurité sanitaire dans les domaines relevant de la compétences des agences (AFSSA, AFSSAPS) et de l'InVS ; d'autre part, deux domaines actuellement mal couverts appellent une réorientation importante. Il s'agit de la santé au travail et des produits chimiques qui n'entrent pas dans le champ de l'AFSSA et de l'AFSSAPS, ainsi que de la sécurité sanitaire de l'environnement ou des milieux de vie. L'AFSSE et ses domaines Créee par la loi du 9 mai 2001, l'AFSSE n'a pas fait l'objet d'une évaluation à l'instar des agences de la loi de 1998. Sa mise en place s'est faite à la fin de l'année 2002 dans des conditions qui lui confèrent une faiblesse inévitable. C'est d'autant plus regrettable que l'ensemble des responsables de cette nouvelle agence ont fait face à la situation et aux nombreuses saisines avec détermination et efficacité (préparation du PNSE - Plan national santé environnement) en 2003-2004 par exemple. Mais le positionnement en effet inadapté de l'AFSSE, la conception erronée de son architecture, ainsi que l'urgence d'une réponse, exigent une première réforme. De très nombreuses et importantes questions sont en cause. Il est donc proposé de regrouper l'évaluation des risques afférents aux milieux de vie dont la santé au travail et les produits chimiques, au sein d'une Agence de Sécurité sanitaire des aliments et des milieux de vie , rapprochement fonctionnel de l'AFSSA et de l'AFSSE ; les règles de fonctionnement et les compétences actuelles de l'AFSSA dans le domaine alimentaire resteraient inchangées. Néanmoins les enjeux environnementaux et les attentes de la société commandent l'ouverture du chantier qui doit permettre de disposer d'une grande Agence de l'Environnement. Dotée de moyens significatifs résultant pour une part des regroupements de services et d'organismes actuels, et d'autre part, de la création de nouvelles unités, elle disposera d'une véritable capacité d'expertise, d'alerte et de préconisation autonomes. Par ailleurs, en complément de cette proposition essentielle, l'évaluation des substances et produits chimiques appelle la constitution d'un groupement d'intérêt public, proposé par les quatre inspections générales. Seule cette structure permettra de faire face à des besoins considérables et de prendre part efficacement aux travaux conduits dans le cadre européen (programme Reach). |
RECOMMANDATIONS
L'ESPRIT DES RECOMMANDATIONS
La loi de juillet 1998 sur la sécurité sanitaire a créé un nouveau dispositif administratif symbolisé par les agences et le renforcement des mécanismes de sécurité sanitaire.
Ces créations ne sont pas dans l'esprit du législateur la marque d'un désengagement de l'Etat.
Au contraire, la nouvelle architecture vise à permettre aux autorités ministérielles un exercice plus efficace des responsabilités de l'Etat dans le domaine de la sécurité sanitaire.
Les propositions qui suivent s'inscrivent dans cette perspective. Elles ont pour objet d'indiquer les améliorations qui s'imposent dans l'architecture générale dont les fondements ne sont pas remis en cause ; certains des principes qu'elle comporte doivent même être renforcés ou mieux concrétisés. Dans ce paysage administratif, l'AFSSE, qui ne fait pas l'objet d'évaluation en elle-même, pose cependant un problème de positionnement qui appelle une proposition de modification.
L'adéquation de la plupart des instances aux objectifs de la loi de 1998 n'exclut pas la nécessité d'améliorer le fonctionnement de ces instances , ainsi que des structures ministérielles qui sont leurs partenaires et dans bien des cas leur tutelle. A ce titre, certains rapports doivent être clarifiés, qu'il s'agisse de l'exercice des contrôles dans la gestion des risques par exemple, ou de la généralisation effective des COM (contrats d'objectifs et de moyens). Le maintien d'une recherche appliquée et d'une expertise de qualité constitue également un objectif essentiel.
Enfin, après avoir indiqué comment les risques actuels doivent être remis en perspective, il convient d'aborder des problèmes nouveaux, aussi qualifiés d' « émergents » , qui appellent des mises en garde et des propositions précises. Ces questions, par définition, n'avaient pas été envisagées en 1998. L'évolution accélérée des réalités mondiales exige maintenant que l'ensemble du dispositif de sécurité sanitaire français, mais aussi européen, puisse y faire face efficacement. Par ailleurs, l'Etat doit avoir une parole forte sur les grandes questions qui interrogent notre société, qu'il s'agisse de l'obésité, d'accidents climatiques ou environnementaux, de l'usage des médicaments, de l'abaissement des contrôles assurant la sécurité sanitaire. Enfin, la difficile question du positionnement de l'expertise dans le domaine des médicaments doit être soulevée sachant qu'elle se pose au niveau mondial.
I. MAINTENIR GLOBALEMENT L'ARCHITECTURE GÉNÉRALE
Le principe général de séparation fonctionnelle entre l'évaluation et la gestion du risque est clairement confirmé , le cas particulier de l'AFSSAPS étant maintenu. Cette séparation a été perçue comme un progrès, confirmé par les événements intervenus depuis 1998.
- il y a donc lieu d'assurer son effectivité et de l'établir dans les domaines actuellement mal couverts comme la santé du travail et les produits chimiques,
- plus précisément, la compétence évaluation du risque de l'AFSSA sur la filière végétale doit devenir effective, conformément à la loi de 1998 qui n'a pas été respectée sur ce point ; cela implique le transfert de la COMTOX et de la CGB à l'AFSSA,
- on retiendra également le maintien de l'ANMV (Agence nationale du médicament vétérinaire) et de l'ensemble des compétences dans ce domaine à l'AFSSA.
Le positionnement inadapté de l'AFSSE, la conception erronée de l'architecture pour les domaines mal couverts déjà évoqués (santé du travail, produits chimiques) ainsi que l'urgence d'une réponse exigent une première réforme. On peut donc proposer le regroupement de l'évaluation des risques afférents à ces domaines au sein d'une agence de sécurité sanitaire des aliments et de l'environnement, rapprochement fonctionnel de l'AFSSA et de l'AFSSE 48 ( * ) . Les règles de fonctionnement et les compétences actuelles de l'AFSSA dans le domaine alimentaire resteraient inchangées. La création d'une grande agence de l'environnement n'est pas récusée a priori mais les délais de mise en oeuvre ne permettent pas de répondre à l'urgence actuelle.
Néanmoins les enjeux environnementaux et les attentes de la société commandent l'ouverture du chantier qui doit permettre de disposer d'une Agence de l'Environnement dans les meilleurs délais. Dotée de moyens significatifs résultant pour une part des regroupements de services et d'organismes actuels, et d'autre part, de la création de nouvelles unités, elle disposera d'une véritable capacité d'expertise, d'alerte et de préconisation autonomes.
La formule du GIP (Groupement d'Intérêt Public), avancée par les quatre inspections générales pour l'évaluation des substances et produits chimiques s'impose compte tenu de l'ampleur des problèmes et de leur extrême diversité. S'y ajoute l'aspect européen, encore plus prégnant ici que dans tous les autres domaines. Cette construction, à la fois juridique et fonctionnelle, vise à regrouper des moyens d'expertise actuellement dispersés tout en maintenant la place de chaque instance intervenante.
Cette formule a d'ailleurs été préfigurée par la saisine conjointe (mars 2004) de l'AFSSA et de l'AFSSE par trois directions ministérielles sur « les troubles des abeilles ».
La distinction entre la veille sanitaire et l'épidémiologie , notamment dans les organismes au sein desquels ces deux objectifs sont coexistants (cas de l'InVS notamment), doit être réaffirmée. Cette distinction est d'autant plus nécessaire que la couverture de nouveaux risques (milieux de vie dont le chimique), exige un partage précis et opérationnel des tâches.
Il sera utile de confirmer au niveau territorial le positionnement des CIRE (cellules interrégionales d'épidémiologie) au sein de l'InVS et le rôle de celui-ci en matière de veille sanitaire. Une répartition opérationnelle des compétences dans le cadre du plan régional de santé publique afin d'éviter toute confusion entre sécurité sanitaire et santé publique sera assurée.
Dès leur mise en place, les compétences des organes consultatifs créés par la loi de santé publique du 9 août 2004 : Haut conseil de santé publique et Comité national de santé publique seront clarifiées par des initiatives visant à :
- Assurer au niveau du fonctionnement de ces organes clarté et réelle cohérence opérationnelle.
- Modifier le cas échéant les dispositions nouvelles qui se révèleraient inadaptées ou insuffisantes sans attendre le verdict de la réalité comme ce fut le cas avec le CNSS.
La mise en place et la « feuille de route » de la nouvelle HAS (Haute autorité de santé), créée par la loi relative à l'assurance maladie du 13 août 2004, ont pour objectif des progrès dans l'appréciation du médicament, dans l'information des praticiens médicaux et la pharmacovigilance. Il convient de s'assurer de l'effectivité des mesures. S'agissant de l'information des médecins sur les médicaments, des avancées sont nécessaires :
- Réalisation complète de la base de données médicaments de l'AFSSAPS (avec le résumé caractéristique du produit) qui aurait dû être acquise depuis plus de deux ans.
- Systématisation d'échanges d'informations (et de coopération) avec la CNAM comme la recherche ponctuelle faite en 2001 sur la cérivastatine en a montré la possibilité. D'une manière générale, implication de la CNAM dans les actions et travaux en matière de pharmacovigilance.
- AFSSAPS : Transférer pour les produits de santé au directeur général la responsabilité de la publication des règles de bonnes pratiques qui sont actuellement validées par le DGS (Directeur général de la Santé).
II. AMÉLIORER LE FONCTIONNEMENT DES INSTANCES
PAR LES CONTRATS D'OBJECTIFS ET DE MOYENS
* Généralisation des COM avec une hiérarchisation des priorités d'objectifs et une actualisation des indicateurs pour tous les aspects de la veille sanitaire.
* Etablissement de liens entre les COM et la structure de la LOLF (loi organique relative aux lois de finances) en s'assurant que la compatibilité avec les cadres de la LOLF ne se fait pas au détriment du caractère opérationnel des COM.
PAR UNE TUTELLE RÉNOVÉE
* Structuration de l'exercice de la tutelle ; distinction entre ce qui relève de la tutelle véritable, de la coopération ou de l'appui scientifique et technique.
- Avant même l'interministériel, s'assurer que l'intra-ministériel fonctionne bien : au sein d'un même ministère la coordination s'impose pour éviter la multiplicité d'interventions auprès d'une même agence.
PAR DES SAISINES MAÎTRISÉES
* Rationalisation de la saisine des agences par les directions ministérielles au moyen de l'établissement de procédures systématisées avec la vérification de la faisabilité en termes de compétences et de moyens.
Cette rationalisation sera particulièrement nécessaire pour le nouveau domaine « environnement » attribué à la nouvelle agence regroupant l'AFSSA et l'AFSSE.
* Structuration des missions interministérielles permanentes à l'instar de ce qui a été engagé dans la préparation du Plan national santé-environnement.
* Efficacité et transparence des plans de contrôle entre agences et administrations ou instances gestionnaires du risque (domaine alimentaire notamment) : établissement ou amélioration de procédures systématisées (protocoles) d'échange d'informations ; méthodologie et résultats des contrôles eux-mêmes.
* Diminution du nombre de saisines de l'AFSSA , notamment par la suppression de l'automaticité de la saisine sur les textes réglementaires nationaux et certaines décisions ponctuelles ; cadrage de l'auto-saisine ; corrélativement extension des saisines de l'AFSSA le plus en amont possible sur les projets de textes ou de décisions européens.
PAR DES COORDINATIONS RENFORCÉES
* Harmonisation et coordination des contrôles entre administrations chargées de la gestion du risque dans le domaine alimentaire (DGCCRF, agriculture y compris services vétérinaires, douanes).
* Mettre un terme à la dispersion de l'expertise en matière d'OGM.
* Au niveau européen pour la sécurité sanitaire des aliments :
-- améliorer l'articulation générale des activités entre l'AESA (Agence européenne de sécurité des aliments) et les agences nationales , notamment à travers le programme de travail de l'Agence européenne, élément nécessaire de coordination entre toutes ces instances ;
-- appliquer pleinement les dispositions prévues dans le règlement par le forum consultatif notamment pour la fonction de réseau ;
-- assurer une coordination dans l'organisation de l'expertise et dans le recrutement des experts, la mutualisation et l'efficacité des travaux d'expertise étant des objectifs essentiels ;
-- rechercher la cohérence des mécanismes d'expertise européen et nationaux, et assurer une clarification dans la confrontation des points de vue scientifiques en cas de désaccord, la procédure d'avis divergent devant être respectée.
PAR UNE EXPERTISE RADICALEMENT REVISITÉE
* L'ensemble de la question
-- Les ressources humaines pour l'expertise posent des problèmes réels dont certains risquent de s'aggraver dans un proche avenir ; la toxicologie fournit l'exemple du domaine dans lequel la crise est la plus grave : le simple maintien du « vivier actuel », déjà insuffisant, est directement menacé . L'insuffisance quantitative de toxicologues aura en outre nécessairement des effets sur la qualité de l'expertise.
-- Le redressement rapide ne pourra s'effectuer dans le domaine des substances et produits chimiques que par la réorganisation permettant la meilleure utilisation des ressources avec la constitution du GIP spécifique ; mais celle-ci ne suffira évidemment pas et n'aurait pas de portée tangible sans ce redressement en terme de moyens .
* Les objectifs principaux
Pour faire face à cette situation, deux objectifs principaux doivent être atteints :
- assurer le recrutement des experts en nombre suffisant à un niveau qualitativement satisfaisant,
- assurer l'autonomie de ces experts face aux diverses formes de pressions ou d'influences susceptibles d'altérer la validité de leurs travaux.
* Les moyens eux-mêmes
Le maintien ou le rétablissement, selon les disciplines, d'une situation satisfaisante passe par plusieurs éléments de réponse, tous indispensables :
-- Rémunérations convenables des fonctions d'expertise par une élévation sensible de leur niveau, ce qui permettra aussi d'éviter de recourir à des experts qui sont trop souvent dans des situations de conflits d'intérêts. Ce qui a été engagé à l'AFSSAPS à cet égard va dans la bonne direction, mais reste nettement insuffisant ; ce relèvement de la rémunération des fonctions d'expertise passe, au-delà des efforts de chaque agence, par une volonté affirmée de l'Etat assortie des moyens nécessaires, perénisée notamment dans le cadre des COM.
-- Maintenir la participation de l'Etat au financement de l'AFSSAPS à un niveau significatif qui corresponde au moins à la couverture des fonctions de contrôle qui relèvent de l'autorité publique.
-- Le statut de l'expertise au sens professionnel, intellectuel et social, doit lui aussi être substantiellement réévalué.
-- La prise en considération de l'expertise par les autorités universitaires et l'ensemble des acteurs de l'enseignement supérieur est essentielle. Actuellement les activités d'expertises ne sont pas valorisées, sauf dans de très rares domaines, et peuvent même constituer un élément négatif dans une carrière d'universitaire ou de chercheur. Il faut que l'expertise soit effectivement valorisée dans les cursus et que son exercice soit facilité dans l'organisation d'ensemble des activités de recherche.
-- L'articulation européenne, voire mondiale : cet objectif est d'autant plus impérieux que dans certains domaines (alimentaire par exemple), une concurrence avec l'échelon européen dans le recrutement des experts pourrait contribuer à affaiblir quantitativement et qualitativement, le flux d'experts au niveau national.
Une initiative innovante : la Haute Autorité de l'Expertise Scientifique
Les témoignages multiples sur les incertitudes du fonctionnement actuel de l'expertise scientifique, illustrées en particulier par les affaires récentes du médicament, appellent une forte initiative .
Il semble souhaitable d'engager la création d'une Haute Autorité de l'Expertise Scientifique . Autonome, chargée d'encadrer l'adaptation à chaque domaine des règles assurant l'indépendance réelle de l'expertise , ainsi que la vérification de leur respect, cette instance nouvelle pourrait être inspirée de la CNIL dans son fonctionnement. Une telle création ne peut résulter que d'une décision législative.
III. FAIRE FACE AUX RISQUES ÉMERGENTS
PAR L'ADAPTATION DES MESURES AUX RISQUES RÉELS ACTUELS
Il convient de porter un éclairage nouveau sur des problèmes préexistants compte tenu de l'évolution des esprits et des données technologiques.
* Ajuster les dispositifs d'identification et de mesure des risques à la gravité de ceux-ci , dans le domaine alimentaire mais aussi dans l'ensemble des champs environnementaux afin que des peurs a priori sans fondement (antennes relais) ou des inquiétudes exagérées (listeria) ne détournent pas des moyens d'analyse et d'actions alors que perdurent des dangers ignorés ou sous-estimés (obésité, exposition aux ultra-violets ...).
Dans cette perspective, l'affaire de l'amiante doit être rappelée . Elle constitue un drame sanitaire majeur avec la perspective de plusieurs milliers de morts par an pendant des années . Elle illustre brutalement la nécessité de tirer les conséquences d'un danger largement avéré depuis longtemps que la pression implacable d'intérêts économiques a dissimulé délibérément. Il n'y avait ni d'inconnue scientifique ni même d'incertitude de degré quant au danger couru . Dans un tel cas, lorsque la certitude l'emporte sur le risque, l'Etat doit être capable d'agir au-delà des pressions de tous ordres.
* Adapter la sévérisation des normes à leur justification scientifique avérée.
* Mieux apprécier la notion de risque zéro , notamment dans les situations où un bénéfice tangible implique inévitablement un risque comme pour les médicaments.
* Informer, éduquer l'opinion et orienter les politiques publiques sur les comportements à risque. L'exemple de la lutte contre le tabagisme illustre combien un objectif qui paraissait inaccessible il y a plus de vingt ans est en passe d'être atteint. L'obésité est aujourd'hui l'un des problèmes les plus graves qui s'ajoute à bien d'autres dont la toxicomanie et l'alcoolisme. Plus ponctuels et moins visibles, d'autres comportements à risque peuvent être identifiés : usage de certains produits chimiques en milieu de travail et domestique, non respect de la chaîne du froid dans le domaine alimentaire ...
PAR L'IDENTIFICATION DES PROBLÈMES NOUVEAUX
a) Dans le domaine sanitaire : renforcer la veille
S'assurer les moyens d'une veille sanitaire adaptée aux risques émergents ou mal identifiés :
* Elaboration d'un schéma national des systèmes d'information pour la veille et l'alerte sanitaire dans l'esprit du « schéma directeur des systèmes d'information » de l'InVS.
* Association des médecins de ville et des établissements non hospitaliers (maisons de retraite, etc) à la veille sanitaire inspiré des réseaux « sentinelles » et plus particulièrement du réseau « grog » pour la grippe.
* Achèvement de la mise en place des dispositifs de transmission en temps réel des informations concernant les motifs d'hospitalisation et les causes de décès.
* Etablissement d'une coordination européenne systématisée au niveau de la veille sanitaire à l'aide notamment du centre de Stockholm ; liaison avec les réseaux existants dans d'autres domaines (« rapex » en matière de sécurité alimentaire par exemple) ou des autorités et administrations à compétences générales (douanes).
b) Dans le secteur alimentaire : identifier et réduire les risques
? Dans le domaine de la sécurité sanitaire des aliments, les risques émergents sont à l'origine des principales inquiétudes que l'on peut identifier aujourd'hui :
-- Les zoonoses virales d'Asie : intervenues au cours des six dernières années, elles illustrent les risques qui peuvent être à l'origine de croisements très dangereux de virus d'origine animale et d'origine humaine. Malgré les progrès substantiels observés dans ce domaine, des efforts devraient être faits dans deux directions : la rapidité de détection des virus animaux et la modélisation prédictive des conditions de leur apparition dans les élevages. De tels travaux ne peuvent efficacement s'envisager qu'à l'échelle européenne.
-- Les biorésistances induites à partir de l'usage d'antibiotiques chez les animaux d'élevage constituent un risque potentiel. L'approfondissement de recherches dans ce domaine correspond donc à une nécessité sanitaire d'autant que la règle de séparation entre antibiotiques à usage animal et à usage humain appliquée dans l'Union européenne ne l'est pas nécessairement dans d'autres régions du monde.
-- L'affaire du « semicarbazide » a montré l'importance de la dimension « technologique » dans la sécurité sanitaire des aliments. Les nouveaux procédés constituent souvent des progrès considérables, notamment dans la perspective d'une sécurité accrue ; mais les risques qu'ils peuvent comporter exigent qu'une attention particulière leur soit apportée tant sous l'angle de la recherche appliquée que sous celui de l'expertise. Un effort particulier doit donc y être consacré.
? L'ouverture des frontières
-- L'insuffisance des moyens de contrôle à l'échelon européen est patent , or c'est à ce niveau que les risques émergents de divers ordres apparaissent ; les nouvelles règles établies par le « paquet hygiène » en 2004 qui permet de déléguer des tâches de contrôle naguère obligatoirement remplies par les services vétérinaires, ne sont pas de nature à susciter la confiance. Mais surtout, l'élargissement de l'Union européenne à dix nouveaux membres pose les problèmes de sécurité sanitaire dans de toutes autres proportions, compte tenu de certains voisinages géographiques. Le nombre de postes de passages aux frontières de l'Union européenne est beaucoup trop élevé si on le compare aux Etats-Unis par exemple, pour que ceux-ci puissent être des lieux de contrôles satisfaisants.
• Il importe donc de renforcer les
contrôles à l'entrée dans l'Union européenne,
notamment par une
pratique significative des prélèvements
pour analyses en laboratoire.
•
Le renforcement des contrôles
dans les nouveaux Etats-membres
s'impose également ; en
effet, les rapports de l'Office alimentaire et vétérinaire de
Dublin et l'analyse de la Commission elle-même en novembre 2003 ont sur
ces sujets fait état d'insuffisances préoccupantes.
• Les moyens de
l'Office alimentaire et
vétérinaire de Dublin
sont déjà
notoirement insuffisants. Malgré l'élargissement à des
nouveaux membres, il n'a pas été envisagé d'augmenter ses
moyens en proportion de cette évolution. Il
est donc
nécessaire de renforcer sensiblement ses effectifs et de focaliser ses
missions
sur les problèmes les plus aigus (respect de la
réglementation dans les nouveaux Etats-membres, effectivité des
contrôles aux frontières communautaires) ;
c) Les enjeux nouveaux de la nutrition
Parmi les compétences de l'AFSSA, l'expertise des propriétés nutritionnelles des aliments expressément prévue a donné à l'Agence l'occasion de se saisir de questions essentielles. Elle n'a pas ici l'exclusivité de la compétence ; il est apparu logique qu'elle exerce cette compétence même si l'une des études (le sel) a pu faire l'objet de critiques. En effet, elle a participé à la prise de conscience du fléau de l'obésité et elle a montré qu'elle joue un rôle irremplaçable.
Il convient donc :
-- de réaffirmer sa compétence dans ce domaine tout en fixant éventuellement davantage le cadre et l'exercice de l'auto-saisine ;
-- de l'encourager dans sa fonction d'appréciation des apports positifs et négatifs d'aliments ou de conditionnements nouveaux à une époque de mondialisation et d'accélération des phénomènes de modes ;
-- de confirmer la nécessité de sa participation à la lutte contre le fléau de l'obésité dans le cadre de ses compétences d'expertise.
S'agissant plus particulièrement de ce problème majeur, l'année qui vient de s'écouler a été marquée par une prise de conscience réelle, mais tardive . Outre deux dispositions sur la publicité, une mesure ponctuelle a été prise avec la suppression des boissons et aliments sucrés dans les distributeurs des établissements d'enseignement secondaire ; or cette mesure a pu être adoptée au cours de la procédure parlementaire après des rebondissements qui se justifient d'autant moins que la cause est évidente face à un tel fléau . La pression doit donc être maintenue notamment par des actions d'éducation pour la santé, par la révision de la situation fiscale de ces produits ou par des recherches sur les comportements alimentaires et la sociologie de l'obésité.
d) Dans le domaine des médicaments : une vigilance globale impérative
* Pour le médicament, la contribution de chaque agence nationale des Etats-membres de l'Union européenne à l'évaluation dans le cadre des procédures d'AMM doit pouvoir être appréciée dans les meilleures conditions de transparence . Il convient donc que les différentes parties prenantes, Agence européenne (EAMA), Commission, gouvernements nationaux et Conseil de l'Union, se donnent les moyens de cette transparence et en assument ensuite l'effectivité dans l'application.
Les risques que la « concurrence » entre agences nationales fait courir à la sécurité sanitaire des produits de santé avec une Union portée à 25 Etats-membres où les demandeurs de l'industrie pharmaceutique pourraient choisir les instances les moins exigeantes ne sont, en effet, pas acceptables, sauf à considérer que les exigences diplomatiques de l'élargissement prévalent sur le niveau de sécurité sanitaire.
La comparabilité des garanties de fiabilité et de sécurité des procédures d'expertises doit être assurée par cette transparence.
* La responsabilité personnelle des experts doit pouvoir être acquise , notamment lorsqu'une expertise a conclu à une absence ou faiblesse de risque alors que des essais cliniques ont fourni des éléments en sens contraire.
* Les efforts faits par l'AFSSAPS sur les déclarations d'intérêts des experts doivent être poursuivis, renforcés, et faire l'objet d'une actualisation permanente systématique. Des sanctions sont à prévoir en cas de manquements à ces obligations et à la déontologie en général.
* Le dispositif français de dispensation du médicament présentant toutes les garanties de sécurité requises , notamment par rapport à certains autres états européens et aux Etats-Unis, il convient d'en maintenir la stricte application . La vente de médicaments par internet n'aurait pas plus de légitimité et de justification que la vente dans les supermarchés. Le caractère spécifique de la dispensation du médicament n'est pas compatible avec un quelconque « dialogue électronique », spécialement à une époque où la iatrogénie médicamenteuse est fortement mise en cause, notamment aux Etats-Unis.
* La banalisation du médicament, au niveau de la publicité cette fois-ci, ne saurait pas être davantage admise, certaines tentatives au niveau communautaire ayant illustré récemment ce risque.
* La contrefaçon des médicaments constitue un problème émergent caractéristique. Actuellement peu important en France, le problème a pris de l'ampleur dans certains pays d'Europe, il est sérieux aux Etats-Unis et constitue un véritable fléau dans de nombreux pays du Tiers-Monde, en Afrique en particulier, la France servant éventuellement de pays de transit. Pour lutter contre cette forme de délinquance, d'autant plus menaçante qu'elle dégage des gains considérables, des initiatives s'imposent au-delà de ce qui a pu être engagé (Conseil de l'Europe, milieux professionnels) :
-- La nécessaire pérennisation de la réglementation assurant le caractère pharmaceutique de la distribution et de la dispensation ;
-- L'établissement d'un réseau de coordination entre les différents acteurs relevant de la puissance publique : AFSSAPS, DGS, Douanes et ce, en liaison avec les instances créées à cette fin ; sur le plan international : Alliance internationale contre les contrefaçons de médicaments associant l'OMS, l'Association Médicale Mondiale, Interpol et l'UNICEF.
-- Au niveau communautaire une prise en compte de cette menace doit également se traduire par l'établissement d'un réseau et d'une instance spécialisée de repérage et d'analyse mais aussi d'action, sachant le caractère international de cette délinquance.
-- Dans cette perspective, il convient que les importations parallèles restent encadrées et que certaines facilités qui ont été données (déconditionnements) à cet égard puissent être remises en cause dès lors que la sécurité sanitaire est menacée et qu'elles constituent une filière de choix pour la contrefaçon.
* Relativement aux essais cliniques et aux procédures d'autorisation de mise sur le marché, l'exigence de transparence s'impose plus que jamais à la lumière des affaires récentes dont celles du Vioxx. L'engagement tout récent pris (6 janvier 2005) par les principales firmes pharmaceutiques en est l'aboutissement tardif. Il revient aux agences et à l'Etat de s'assurer de l'effectivité et de la pérennité de ces engagements.
-- Au-delà, c'est toute une logique d'orientation de la recherche de nouveaux médicaments qui est en cause. Il convient de ne pas favoriser de fausses innovations pour lesquelles des pratiques de marketing et d'agressivité financière sont déployées sans vergogne (propositions de prix d'appel en milieu hospitalier et exigences parallèlement très élevées pour le prix public remboursable par l'assurance-maladie). Une transparence, intersecteurs et internationale s'impose ici aussi.
La réorientation des recherches vers des maladies moins répandues ou moins immédiatement solvables constitue un objectif majeur dans le domaine des produits pharmaceutiques.
* Les erreurs d'appréciation et les orientations contestables observées à partir de cas d'expertises de certains médicaments depuis quelques années au niveau mondial et européen, exigent qu'une analyse de cette problématique puisse être réalisée : la crise de confiance dans les médicaments provoquée par l'affaire du VIOXX doit être l'occasion d'un sursaut collectif. Il est donc souhaitable que le Parlement s'en saisisse par l'une de ses procédures de contrôle ou d'évaluation : mission d'information ou rapport de l'OPECST .
______
EXAMEN DU RAPPORT PAR L'OFFICE
L'Office a procédé à l'examen du rapport de M. Claude Saunier, sénateur, rapporteur de l'étude sur « L'application de la loi n° 98-535 du 1er juillet 1998 relative au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme ».
M. Claude Saunier, sénateur, rapporteur, a rappelé que ce rapport correspond à une saisine de droit prévue dans la loi de 1998 (art. 30) qui dispose que l'Office parlementaire d'évaluation des choix scientifiques et technologiques et le Gouvernement procèdent à l'évaluation de l'application de la loi dans un délai de cinq ans après son entrée en vigueur. Il s'agissait donc a priori d'un rapport d'analyse et de constat dans une perspective administrative, juridique et technique. Or des événements qui se sont produits dans différents domaines au cours des dernières années ont attiré l'attention sur des aspects factuels qui ont pris des proportions imprévues : la canicule, les interrogations sur les produits phytosanitaires (abeilles), et tout récemment, le médicament au niveau mondial.
Dès lors, il convenait d'en tenir compte dans l'appréciation des instances et mécanismes prévus par la loi de 1998.
Le rapporteur a ensuite présenté un état des lieux de la sécurité sanitaire dans son ensemble. La nouvelle organisation a pour origine une proposition de loi de M. Claude Huriet, alors sénateur ; elle comporte des acquis substantiels et les principes sur lesquels elle repose ont été validés par l'expérience : séparation de l'évaluation et de la gestion du risque dans le domaine alimentaire, refus d'une agence unique générale du type de la Food and Drug Administration (FDA) américaine.
Des interrogations se posent toutefois sur la mise en oeuvre de références telles que le « risque zéro » et le principe de précaution dès lors que l'on observe, d'une part, de fausses inquiétudes, et, d'autre part, le maintien, voire la croissance de comportements à risque dont l'obésité est le plus marquant par sa gravité, mais qui est loin d'être le seul.
S'agissant de l'Agence française de sécurité sanitaire des aliments (AFSSA), le rapporteur a fait part de son appréciation globale très positive de la mise en place de l'Agence, d'autant que celle-ci a eu lieu dans le contexte de « séisme » de l'année 1999 avec l'ESB (« vache folle »). L'intégration des laboratoires du Centre national d'études vétérinaires et alimentaires (CNEVA) lui a donné la masse critique pour remplir efficacement la plupart de ses missions et la Direction de l'évaluation des risques nutritionnels et sanitaires (DERNS) exerce de son côté avec efficacité ce qui constitue le « coeur de métier » de l'Agence. La crédibilité à l'égard de tous les partenaires et intervenants du domaine semble acquise et vérifiée.
La séparation entre l'évaluation et la gestion du risque est généralement respectée. La maîtrise du volume des saisines de l'Agence doit être assurée. Le non respect de la compétence de l'Agence sur les produits phytosanitaires, d'une part, et la multiplicité des instances d'expertise sur les OGM, d'autre part, constituent des difficultés qui doivent être levées en réglant le problème de la valorisation et du statut de l'expertise.
Traitant ensuite de l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS), le rapporteur a rappelé la lourde mission qui est celle de l'Agence : l'évaluation et la gestion du risque pour tous les produits de santé.
Après une montée en puissance difficile, au cours de laquelle l'ensemble des fonctions de base de l'Agence (procédures d'autorisation de mise sur le marché notamment) ont toujours été assurées de manière satisfaisante, la stabilisation a été acquise. De nombreuses réformes engagées en dehors de l'Agence peuvent remettre en cause cette stabilisation, le transfert de la commission de la transparence des médicaments à la nouvelle Haute Autorité de Santé (HAS) étant la plus récente d'entre elles.
Si l'AFSSAPS assure l'ensemble de ses fonctions, notamment dans des domaines nouveaux (dispositifs médicaux, cosmétiques), elle est toutefois interpellée comme l'ensemble des agences chargées du médicament dans le monde, par le véritable séisme déclenché par la crise du Vioxx depuis le 30 septembre 2004.
Le retrait de cet anti-inflammatoire non stéroïdien par le laboratoire Merckx s'est fait de façon abrupte, le souci prioritaire étant l'information des autorités boursières et non sanitaires. Dès lors, le problème de la connaissance ou de la dissimulation par le fabricant de tests cliniques révélant le risque excessif du médicament s'est posé avec acuité. Un chiffre de 28.000 décès cardiaques entraînés par le Vioxx pour les seuls Etats-Unis a été avancé. La FDA américaine est fortement mise en cause. L'Agence européenne du médicament elle-même n'a pas procédé à la réévaluation qu'elle aurait dû faire et que l'AFSSAPS lui avait demandée. La décision de publication des essais cliniques annoncée par l'ensemble de l'industrie pharmaceutique mondiale au début du mois de janvier 2005 est la suite logique, mais tardive, de cette crise majeure.
La iatrogénie médicamenteuse trouve là une illustration brutale. Il faut en tirer la leçon : des efforts réels à entreprendre, notamment une meilleure structuration de la pharmacovigilance.
Par ailleurs, un risque émergent qui pour le moment n'a pas d'impact en France ne doit pas être négligé : c'est celui de la banalisation du médicament, notamment à travers la vente par Internet, le cas américain fournissant une référence inquiétante, qui est malheureusement suivie depuis très peu de temps par l'Allemagne.
En outre, la dimension européenne des procédures d'autorisation de mises sur le marché dans une Europe à 25 ne devra pas avoir pour conséquence un abaissement de la sécurité atteinte aujourd'hui.
Traitant des clarifications à opérer dans l'ensemble de l'organisation de la sécurité sanitaire, M. Claude Saunier, sénateur, rapporteur, a indiqué que l'AFSSE -- exclue de la présente évaluation en raison de sa trop récente mise en place -- ne peut rester dans un positionnement incertain. Il conviendrait donc de la regrouper avec l'AFSSA, l'objectif restant celui de la création d'une véritable agence de l'environnement aux compétences et aux moyens bien déterminés, agence qui regrouperait tout ce qui oeuvre actuellement dans ces domaines. L'analyse des risques pour la santé au travail et les produits chimiques, qui est tout à fait insuffisante, devrait enfin être réalisée.
En conclusion de son rapport, M. Claude Saunier, sénateur, rapporteur, a présenté plusieurs recommandations. Outre le regroupement de l'AFSSA et de l'AFSSE, le rapporteur a souligné l'importance de la création d'une Haute autorité de l'expertise scientifique. Autonome, chargée d'encadrer l'adaptation à chaque domaine des règles assurant l'indépendance réelle de l'expertise, ainsi que la vérification de leur respect, cette instance nouvelle pourrait être inspirée de la Commission nationale informatique et liberté (CNIL) dans son fonctionnement.
D'autres recommandations visent également l'expertise scientifique à travers le statut et la rémunération des experts ainsi que la valorisation de leur fonction. Parmi plusieurs recommandations relatives au médicament, il a particulièrement signalé la nécessité de transparence des processus d'autorisation de mise sur le marché (AMM) au niveau européen et l'exigence d'une preuve de réelle innovation lors de la demande d'autorisation.
Le rapporteur a aussi recommandé que le Parlement se saisisse des questions posées depuis quelques années au niveau mondial par le médicament à travers une mission d'information ou un rapport de l'Office.
M. Paul Blanc, sénateur, a évoqué le dépôt récent d'une proposition de résolution portant création d'une commission d'enquête au Sénat sur le médicament.
M. Pierre Laffitte, sénateur, a souligné la justesse de l'analyse faite par le rapporteur sur les problèmes fondamentaux de l'expertise qui s'observent dans tous les domaines et dans tous les pays d'Europe. C'est particulièrement regrettable pour la France qui a longtemps eu de nombreux experts de valeur ; il s'agit là d'un problème sociétal complexe.
M. Claude Birraux, député, Premier-vice président, a estimé qu'à l'origine la création des agences avait été la marque d'un échec des administrations centrales, l'exemple de la direction générale de la santé étant particulièrement éclairant. L'épidémiologie devait depuis longtemps être une priorité, ce qui n'est pas le cas, et l'essentiel reste à faire pour la pharmacovigilance.
Mme Marie-Christine Blandin, sénateur, a souligné la grande faiblesse de moyens des directions départementales de l'action sanitaire et sociale (DDASS) pour procéder aux analyses et aux contrôles nécessaires. Elle a remarqué que le domaine du phytosanitaire n'était pas surveillé, alors que le maniement et le dosage de ces produits dans de nombreuses situations, y compris par les services municipaux, ne sont pas correctement effectués. Elle a souhaité que les problèmes de l'expertise et de l'épidémiologie ne soient pas oubliés dans la future loi sur la recherche.
M. Henri Revol, sénateur, président, a salué ce travail d'évaluation du rapporteur et rappelé l'origine législative de la saisine.
L'Office a adopté, à l'unanimité des membres présents, le rapport « sur l'application de la loi n° 98-535 du 1er juillet 1998 relative au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme » ainsi que l'ensemble des recommandations proposées par le rapporteur.
______
LISTE DES PERSONNES AUDITIONNÉES
Par M. Claude SAUNIER (17 novembre 2004 - 8 décembre 2004)
-- Mme Fabienne BARTOLI, Conseiller technique pour les industries de santé - Cabinet de M. Douste-Blazy, Ministre des solidarités, de la santé et de la famille
-- M. Gilles BRÜCKER, Directeur général de l'Institut de veille sanitaire
-- Pr Charles CAULIN, Professeur de médecine à l'hôpital Lariboisière à Paris ; ancien président de la commission d'autorisation de mise sur le marché
-- M. Guillaume CERUTTI, Directeur général de la concurrence, de la consommation et de la répression des fraudes
-- Dr Yves COQUIN, Directeur adjoint au Directeur général de la Santé, Chef du service prévention, programmes de santé et gestion des risques,
-- Dr William DAB, Directeur général de la Santé
-- M. Pierre DELOMÉNIE, Inspecteur général des Affaires Sociales
-- Mme Monique ELOIT, Directrice auprès du Directeur général de l'AFSSA
-- M. Martin HIRSCH, Directeur général de l'AFSSA
-- Pr Claude HURIET, Sénateur honoraire, rapporteur de la mission sénatoriale d'information sur la sécurité sanitaire en 1997 - co-auteur de la proposition de loi devenue la loi du 1 er juillet 1998. Président de l'Institut Curie
-- M. Jean MARIMBERT, Directeur général de l'AFSSAPS
-- M. Thierry MICHELON, Ingénieur en chef des mines - sous-directeur de la gestion des risques des milieux au ministère de la santé
-- M. Pierre de MONLIVAUT, Conseiller technique au Cabinet de M. Douste-Blazy, Ministre des solidarités, de la santé et de la famille
-- M. Gérard PASCAL, Directeur de recherche honoraire à l'INRA
-- Pr Roland SAMBUC, chargé du pôle santé publique et sécurité sanitaire - Cabinet de M. Douste-Blazy, Ministre des solidarités, de la santé et de la famille
-- M. Bruno TOUSSAINT, Directeur de la rédaction de la revue Prescrire
-- Mme Emmanuelle WARGON, Adjointe au Directeur général de l'AFSSAPS)
par M. Bernard SEILLIER (30 septembre 2003 - 10 juin 2004)
en FRANCE
-- M. Olivier ANDRAULT, Confédération de la consommation, du logement et du cadre de vie - CLCV
-- M. Christian BABUSIAUX, Conseiller-maître à la cour des Comptes, ancien président du Conseil National de l'Alimentation - ancien Directeur général de la concurrence, de la consommation et de la répression des fraudes
-- M. Jérome BÉDIER, Président de la Fédération du commerce et de la distribution
-- Mme Aline BESSIS-MARAIS, Responsable affaires publiques du LEEM - Les entreprises du médicament
-- M. Franck BONNEVAL, Membre du bureau de Jeunes Agriculteurs
-- M. Jean-André BOUCHAND, Directeur des laboratoires de la DGCCRF
-- M. Guillaume BOURGE, Jeunes Agriculteurs
-- M. Gilles BRÜCKER, Directeur général de l'Institut de veille sanitaire
-- Pr Charles CAULIN, Professeur de médecine à l'hôpital Lariboisière à Paris ; ancien président de la commission d'autorisation de mise sur le marché
-- Dr CHABRIER, Président du Syndicat national des médecins spécialistes en endocrinologie, diabète, maladies métaboliques et nutrition
-- M. Michel CHASSANG, Président de la Confédération des syndicats médicaux français (CSMF)
-- M. Claude CHEREAU, Inspecteur général de l'agriculture
-- Mme Christine CHERBUT, Centre de recherche Nestlé, ancien expert à l'INRA et à l'AFSSA
-- M. François COINDREAU, Association nationale des industries alimentaires (ANIA) - président de la commission qualité-sécurité
-- Mme Dominique COMBRET, Présidente de l'Association des diététiciens de langue française
-- M. Yves COQUIN, Directeur adjoint au directeur général de la Santé, chef du service prévention, programmes de santé et gestion des risques
-- M. Patrick DEHAUMONT, Directeur de l'Agence nationale du médicament vétérinaire à Fougères (AFSSA)
-- M. Pierre DELOMÉNIE, Inspecteur général des Affaires Sociales
-- M. Noël DIRICQ, Chef du service de la régulation et de la sécurité à la DGCCRF
-- M. Christian DUBREUIL, Inspecteur général de la santé publique, vétérinaire
-- M. Jean-Pierre d'ESTIENNE D'ORVES, Association nationale des industries alimentaires (ANIA)
-- Mme Monique ELOIT, Directrice auprès du directeur général de l'AFSSA
-- M. Pierre FEILLET, Directeur de recherche émérite à l'INRA
-- Pr Marc FELLOUS, Président de la Commission du Génie Biomoléculaire
-- Mme Michèle FROMENT-VÉDRINE, Directrice générale de l'AFSSE
-- Pr FLAHAUT, Expert auprès de la mission des inspections générales, professeur des universités et praticien hospitalier épidémiologiste (INSERM)
-- M. Frédéric GARD, Confédération des syndicats médicaux français
-- M. Hervé GAYMARD, Ministre de l'Agriculture, de l'Alimentation, de la Pêche et des Affaires rurales
-- M. Michel GAGNEUX, Inspecteur général des Affaires Sociales
-- M. Thierry GESLAIN, Directeur qualité et consommateurs à l'Association nationale des industries alimentaires (ANIA)
-- M. Philippe GUERIN, Président du Conseil national de l'alimentation
-- M. Patrick HERVÉ, Président de l'Etablissement Français du Sang
-- M. Martin HIRSCH, Directeur général de l'AFSSA
-- Pr Claude HURIET, Sénateur honoraire, rapporteur de la mission sénatoriale d'information sur la sécurité sanitaire en 1997 - co-auteur de la proposition de loi devenue la loi du 1 er juillet 1998. Président de l'Institut Curie
-- M. Christian JACOB, Ministre délégué aux petites et moyennes entreprises, au commerce et à l'artisanat, aux professions libérales et à la consommation
-- Pr Damien JOLLY, Professeur de santé publique au CHU de Reims - Président de l'Observatoire régional de la santé Champagne-Ardennes
-- M. Michel JOLY, Vice-président de Jeunes Agriculteurs
-- M. Thierry KLINGER, Directeur général de l'Alimentation au ministère de l'Agriculture
-- M. Philippe LAMOUREUX, Directeur général de l'Institut national de prévention et d'éducation pour la Santé (INPES)
-- M. Jean-Paul LAPLACE, Président de l'Institut français pour la nutrition et directeur de recherche à l'INRA
-- Mme Catherine LASSALLE, Directeur des affaires scientifiques pharmaceutiques et médicales au LEEM - Les entreprises du médicament
-- Mme Roselyne LECOURT, Chargée de mission à la DGCCRF pour les questions européennes et internationales
-- M. Yves LEFORBAN, Inspecteur général de la santé publique vétérinaire
-- M. Bernard LEMOINE, Vice-président délégué du LEEM - Les entreprises du médicament
-- Pr Claude LE PEN, Professeur d'économie de la santé Paris-Dauphine
-- Pr Pierre LOUISOT, Professeur à la Faculté de médecine de Lyon-sud - biochimie (INSERM)
-- M. Jean MARIMBERT, Directeur général de l'AFSSAPS
-- Pr Ambroise MARTIN, Directeur de la direction de l'évaluation des risques nutritionnels et sanitaires (AFSSA)
-- M. Benoît MANGENOT, Directeur général de l'Association nationale des industries alimentaires (ANIA)
-- M. Jean-François MATTEI, Ministre de la Santé, de la Famille et des Personnes handicapées
-- M. MARTEAU, Membre du Bureau national de la FNSEA, chargé de l'agro-alimentaire
-- Pr Daniel MARZIN, Président de la commission d'étude de la toxicité des produits antiparasitaires à usage agricole et des produits assimilés (COMTOX)
-- Pr Joël MÉNARD, Professeur de médecine, ancien Directeur général de la santé
-- M. Bernard MOINIER, Comité des salines de France
-- M. Dominique MOUROT, Directeur adjoint de l'Agence nationale du médicament vétérinaire à Fougères (AFSSA)
-- Mme MORAUT, Union fédérale des consommateurs - Que Choisir ?
-- M. NAIRAUD, Secrétaire général du Conseil national de l'alimentation
-- M. Gérard PASCAL, Directeur scientifique pour la nutrition humaine et la sécurité des aliments à l'INRA
-- Mme Aline PEYRONNET, Sous-directrice Protection du consommateur à la DGCCRF
-- Mme Dorothée QUICKERT-MENZEL, Confédération de la consommation, du logement et du cadre de vie
-- M. Etienne RECHARD, Directeur entreprises de COOP de France (Confédération française de la coopération agricole)
-- M. Pascal SANDERS, Directeur du laboratoire d'études et de recherches sur les médicaments vétérinaires et les désinfectants (Agence nationale du médicament vétérinaire - AFSSA)
-- M. Maxime SCHWARTZ, Directeur de la programmation et des laboratoires (AFSSA)
-- M. Michel SETBON, Expert auprès de la mission des inspections générales, directeur de recherche CNRS (Aix-en-Provence), sociologue
-- M. Jean-Pierre TILLON, Directeur scientifique de la Confédération française de la coopération agricole
-- M. Paul VIALLE, Président du conseil d'administration de l'AFSSA
-- Mme Catherine VIGREUX, Industrielle (société Roquette), membre du conseil d'administration de l'Institut français pour la nutrition
-- M. Laurent VACHEY, Inspecteur des finances
-- M. Philippe VERGER, Directeur de recherche à l'INRA, membre de comités d'experts (AFSSA, Union européenne, FAO)
-- Mme Emmanuelle WARGON, Adjointe au Directeur général de l'AFSSAPS
à BRUXELLES
-- M. Laurent BOCHEREAU, Chef de l'Unité « sécurité des systèmes de production alimentaire » à la Commission européenne
-- M. Christian MASSET, Représentant permanent adjoint de la France auprès de l'Union européenne
-- M. Geoffrey PODGER, Directeur exécutif de l'Autorité européenne de Sécurité des Aliments (AESA)
-- M. Robert VANHOORDE, Chef de l'Unité « Relations avec l'Autorité européenne de Sécurité des Aliments » à la Commission européenne
-- M. Eric ZUNINO, Conseiller pour la Sécurité alimentaire à la Représentation permanente de la France
au ROYAUME-UNI
-- Dr BELL, Directeur exécutif de l'Agence de sécurité des aliments
-- Mme Lis BIRRAINE, Directrice de la communication de l'Agence de protection de la santé
-- Pr BRECKENRIDGE, Président exécutif de l'Agence (britannique) de réglementation des médicaments et des produits de santé
-- Dr Françoise CLUZEAU, Responsable des relations extérieures du National institute for clinical excellence (NICE)
-- M. Henry DERWENT, Directeur Climat, énergie et risques environnementaux du Ministère de l'environnement, de l'alimentation et des affaires rurales
-- M. Sian GRIFFITHS, Membre du conseil de direction de l'Agence de la protection de la santé ; président de la faculté de santé publique
-- Dr Paul HARRISSON, Directeur de l'Institut pour l'environnement et la Santé
-- Dr Ian HUPSON, Division des autorisations de l'Agence (britannique) de réglementation des médicaments et des produits de santé
-- M. Patrick LE COURTOIS, Chef de l'unité de pré-autorisation à l'EMEA - European Agency for the Evaluation of Medicinal Products ; Agence européenne d'évaluation des médicaments
-- Dr Paul LEINSTER, Directeur de la protection environnementale à l'Agence de l'environnement
-- Dr Gordon MUNRO, Chef du département médicaments de l'Agence (britannique) de réglementation des médicaments et des produits de santé
-- M. Angus NICOLL, Directeur de la surveillance des maladies contagieuses à l'Agence de protection de la santé
-- Pr Michael RAWLINS, Président du NICE (National Institute for Clinical Excellence)
-- Dr Angela ROBINSON, Directeur médical du Service national du sang
-- Dr Pat TROOP, Directrice générale de l'Agence de protection de la santé
-- Dr Louise WOOD, Responsable des bases de données à l'Agence (britannique) de réglementation des médicaments et des produits de santé
______
* 1 Les rapporteurs de l'OPECST ont été M. Alain Claeys, député et M. Claude Huriet, sénateur.
* 2 Respectivement p. 19 et 18
* 3 La sécurité sanitaire - Didier Tabuteau chez Berger Levrault Roger 39 (2 ème édition) ; l'auteur a dirigé l'Agence du Médicament de 1993 à 1997 et a été directeur de cabinet de M. Bernard Kouchner, Ministre de la Santé
* 4 Rapport précité p. 42.
* 5 Rapport de la Cour des Comptes Septembre 2002 en application des lois de financement de la sécurité sociale.
* 6 InVS rapport annuel 2002.
* 7 Les agences sanitaires sous la tutelle : le nouveau paysage sanitaire impulsé en 1998 et les interactions des agences entre elles - document de travail 25 avril 2002.
* 8 Op.cité page 67.
* 9 Cf. supra - pour le COM de l'InVS et les remarques de la Cour des Comptes sur la mise en oeuvre de ce nouvel outil avec les agences en général.
* 10 Commission d'enquête sur « les conséquences sanitaires et sociales de la canicule » AN XIIème législature - rapport n° 1455 Tome 2 - page 208.
* 11 Cf. rapport Claude Huriet sur la proposition de loi - Sénat n° 413 (1996-1997).
* 12 Auditions publiques de la commission des affaires sociales du Sénat mai 2000 n° 445 (1999-2000) p.8.
* 13 OPECST Rapport n° 346 (AN XIIè Législature) n° 52 Sénat (2002-2003) du 6 novembre 2002 (pages 99 et 111).
* 14 Le principe de précaution s'applique dans des conditions particulières, ainsi que l'a rappelé la Commission européenne : COM (2000) - Communication de la commission sur le recours au principe de précaution.
« le principe de précaution (...) couvre les circonstances particulières où les données scientifiques sont insuffisantes, peu concluants ou incertaines, mais où, selon des indications découlant d'une évaluation scientifique objective et préliminaire, il y a des motifs raisonnables de s'inquiéter que les effets potentiellement dangereux sur l'environnement et la santé humaine,animale ou végétale soient incompatibles avec le niveau choisi de protection.
(...) Le recours au principe de précaution présuppose :
- l'identification d'effets potentiellement négatifs découlant d'un phénomène, d'un produit ou d'un procédé ;
- une évaluation scientifique du risque qui, en raison de l'insuffisance des données, de leur caractère non concluant ou encore de leur imprécision, ne permet pas avec une certitude suffisante d'estimer le risque en question ».
* 15 Communiqué de presse InVS-AFSSA du 10 mai 2004 « Estimation de l'importance des infections d'origine alimentaire en France ».
* 16 La situation est à peu près comparable au Royaume-Uni où les autorités ont fait de la lutte contre les salmonelloses une priorité, notamment dans le secteur de la volaille.
* 17 In La Recheche Février 2001
* 18 Ayant participé à l'enquête sur l'accident de SEVESO, le Pr. TUBIANA a précisé à ce sujet : « Je peux vous dire qu'il n'y a pas eu de catastrophe, seulement trente avortements entraînés par une panique injustifiée. Les foetus étaient normaux, et on a sacrifié trente bébés pour rien.
(cité par Pierre KOHLER, L'imposture verte - Albin Michel - septembre 2002)
* 19 La sécurité de l'alimentation - Travaux du comité scientifique de la FCD (Fédération des entreprises du commerce et de la distribution) Septembre 2001.
* 20 P-H BOURRELIER, ingénieur général des mines, coordonateur du rapport de l'Académie des Sciences sur « la contamination des sols par les éléments en trace ».
* 21 Le CNA, Conseil National de l'Alimentation, est un organisme consultatif créé par décret du 27 novembre 1985 et placé auprès des ministères de l'agriculture, de la santé et de la consommation.
Composé de 47 membres, il réunit tous les partenaires de la filière agro-alimentaire : consommateurs, professionnels du secteur et de la distribution et experts qualifiés.
Selon les informations portées sur son site internet, il ne se substitue pas aux instances qualifiées en matière scientifique. Il a pour vocation, aux termes du décret de 1985, d'émettre des avis, sur consultation des ministres, sur les questions se rapportant à la politique gouvernementale en matière alimentaire, qu'il s'agisse d'hygiène, de qualité ou de nutrition. Il formule des appréciations socioéconomiques sur la gestion des risques
* 22 Publié chez Albin Michel - mars 2002 - pages 215-225
* 23 Cité par M. HIRSCH ibidem Page 184.
* 24 Canicules - novembre 2003 Fayard Editeur.
* 25 Cf. supra 1 ère partie du présent rapport.
* 26 Cf. supra page 78
* 27 On rappellera que seule la DGCCRF parmi les trois tutelles a accepté de participer à ces travaux.
* 2 La recherche : missions et politique de l'AFSSA - orientations 2002/2005, page 7.
* 28 Cité par P. FEILLET in « Le bon vivant » - INRA Editions février 2002
* 29 Le rapporteur des quatre projets de textes, M. Horst Schnellhardt avait d'ailleurs argumenté au sujet de la participation alors envisagée des personnels des abattoirs pour les porcs et les veaux d'engraissement que cette participation aux contrôles officiels serait contre-productive car « cela ne contribuera pas à l'amélioration de la sécurité alimentaire, ni ne répondra aux exigences d'un contrôle indépendant ».
* 30 Renforcer la sécurité sanitaire en France - Sénat 1996-1997 n° 196 page 22.
* 31 Art. 5322-2 du Code de la santé publique.
* 32 Rapport d'audit IGF/IGAS sur l'AFSSAPS - Décembre 2002 : rapport de synthèse page 62.
* 33 Rapport d'audit précité page 43
* 34 Rapport établi en mars 1998 par le Pr Patrice Quéneau, professeur de thérapeutique, médecin des hôpitaux CHU de St Etienne, Président de l'association pédagogique nationale pour l'enseignement de la thérapeutique.
* 35 Enquête américaine conduite par la Women's Health Initiative et en Grande-Bretagne sous le nom de Women Million Study.
* 36 Outre les AINS « classiques » utilisés comme anti-inflammatoires ou antalgiques, cette classe pharmacologique comprend également l'acide acétylsalicylique (aspirine) quand il est utilisé à des doses supérieures à 500 mg/jour.
* 37 Cité dans Le Figaro du 11 novembre 2004 par M. J.M. Bader.
* 38 Le Monde du 25 janvier 2005.
* 39 Le Figaro (25 janvier 2005).
* 40 Données communiquées par M. Pierre Delval, cabinet du ministre de l'économie et des finances au colloque sur les contrefaçons du médicament organisé le 3 novembre 2004 par l'Ordre national des pharmaciens ; la plupart des éléments chiffrés dont il est fait état ici ont été communiquées par les divers intervenants lors de ce colloque.
* 41 Cité par le Figaro le 5 novembre 2004.
* 42 F. Klein, « @-Santé aux Etats-Unis : le cyber Far West », ACIP Magazine, n° 205-206, p. 12.
* 43 L. Silbert, « L'Etat du Michigan donne un coup d'arrêt à la vente de médicaments sur Internet », Le Quotidien du pharmacien, n° 1856, 24 janvier 2000, page 12.
* 44 Art L.1311-1 du CSP : Sans préjudice de l'application de législations spéciales et des pouvoirs reconnus aux autorités locales, des décrets en Conseil d'Etat, pris après consultation du Haut Conseil de la santé publique et, le cas échéant, du Conseil supérieur de la prévention des risques professionnels, fixent les règles générales d'hygiène et toutes autres mesures propres à préserver la santé de l'homme, notamment en matière :
- de prévention des maladies transmissibles ;
- de salubrité des habitations, des agglomérations et de tous les milieux de vie de l'homme ;
- d'alimentation en eau destinée à la consommation humaine ;
- d'exercice d'activités non soumises à la législation sur les installations classées pour la protection de l'environnement ;
- d'évacuation, de traitement, d'élimination et d'utilisation des eaux usées et des déchets ;
- de lutte contre les bruits de voisinage et la pollution atmosphérique d'origine domestique ;
- de préparation, de distribution, de transport et de conservation des denrées alimentaires.
* 45 Il Il s'agit là notamment de ce que l'InVS appelle « la mise en chantier de la surveillance d'événements non spécifiques à partir des services d'urgence et de veille sur les données de mortalité émises par l'INSEE » (ibidem page 49)
* 46 REACH : Registration, Evoluatoin, Authorization of Chemicals.
* 47 Banque nationale des cas et des demandes d'information toxicologiques des centres anti-poison, données toxicologiques du réseau national de vigilance des pathologies professionnelles des centres de pathologie professionnelle, réseau de toxico-vigilance agricole de la Mutualité sociale agricole
* 48 Ou « agence de sécurité sanitaire des milieux de vie et des produits alimentaires » proposé par le rapport des quatre inspections générales.