







CHAPITRE
III
GARANTIR ET SÉCURISER L'ACCÈS DES FRANÇAIS
AUX MÉDICAMENTS DU QUOTIDIEN ET AUX PRODUITS DE SANTÉ
INNOVANTS
Article
31
Réforme du financement de l'établissement français
du sang (EFS)
Cet article propose de faire évoluer le modèle de financement de l'Établissement français du sang (EFS) vers un modèle mixte. D'une part, il prévoit que le tarif de cession des produits sanguins labiles est fixé en tenant compte de divers coûts de revient. D'autre part, il pérennise une dotation annuelle versée par l'assurance maladie à l'EFS.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - L'EFS connaît des difficultés de fonctionnement chroniques qui se sont aggravées et mettent en jeu la viabilité de son modèle économique
A. Des difficultés de fonctionnement de l'EFS aiguisées par l'évolution de son environnement
1. L'organisation de l'EFS : un héritage de réformes successives dans un environnement complexe
L'organisation actuelle de la filière sang résulte des réformes réalisées en 1993 à la suite de la crise dite du « sang contaminé ». Les missions auparavant réunies au sein du Centre national de transfusion sanguine (CNTS) ont alors été réparties entre trois opérateurs :
- l'Agence française du sang, remplacée en 2000 par l'Établissement français du sang (EFS), disposant du monopole de la collecte du sang et de certains de ses dérivés ;
- le Laboratoire français du fractionnement et des biotechnologies (LFB), disposant du monopole pour le fractionnement du sang, c'est-à-dire la production de médicaments issus de certaines de ses composantes (par exemple pour traiter l'hémophilie) ;
- l'Institut national de la transfusion sanguine (INTS), qui menait alors une activité de recherche et de formation et hébergeait notamment le Centre national de référence pour les groupes sanguins (CNRGS). En 2021, le CNRGS et le département « enseignement et formation » de l'INTS ont rejoint l'EFS.
Enfin, l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) assure la responsabilité de la sécurité sanitaire des produits issus du sang.
L'EFS a effectué 2,7 millions de prélèvements de sang total, de plasma et de plaquettes auprès de plus de 1,5 million de donneurs en 2022937(*).
L'EFS produit et fournit :
- des produits sanguins dits « labiles » (PSL) destinés à la transfusion938(*) et cédés aux établissements de santé ; il s'agit des concentrés de globules rouges, des plaquettes et du plasma dit « thérapeutique » ;
- le plasma pour fractionnement destiné au LFB, servant à produire des médicaments dérivés.
L'EFS dispose d'un monopole pour une partie seulement des
PSL
- les concentrés de globules rouges et les plaquettes
(à l'exclusion donc du plasma dit
« thérapeutique ») - ainsi que pour le plasma
pour fractionnement. Les tarifs de ces produits sont fixés par
arrêté ministériel. En revanche, depuis un arrêt
du Conseil d'État du 23 juillet 2014, les prix du plasma
cédé à des fins thérapeutiques relèvent du
secteur concurrentiel939(*).
Dans son rapport public annuel de 2019, la Cour des comptes indiquait que depuis 2010, « le contexte économique et règlementaire de la filière française du sang et des produits dérivés du sang avait profondément évolué et emportait des risques significatifs pour sa pérennité »940(*). L'ouverture à la concurrence du plasma thérapeutique a contribué à la fragilisation de l'EFS, l'obligeant à baisser les prix pratiqués pour conserver ses parts de marché.
S'agissant de l'organisation de l'EFS, la Cour soulignait notamment :
- la croissance problématique des dépenses de personnel, liée à un recours important à l'intérim compte tenu de la difficulté à fidéliser certains personnels ;
- la faible productivité des collectes dont le pilotage national se révèle insuffisant, conduisant au maintien de petites collectes (30 à 50 dons) qui mobilisent pourtant des moyens importants.
Enfin, la directive UE 2016/1214 du 25 juillet 2016 relative aux normes et spécifications techniques applicables aux établissements de santé, en vigueur depuis 2018, imposait de nouvelles contraintes supposant pour l'EFS de réaliser des investissements et de revoir son organisation.
2. Un modèle économique structurellement déséquilibré
Le modèle historique de financement de l'EFS repose sur les recettes issues des cessions de PSL aux établissements de santé et au LFB, qui représentent 90 % du chiffre d'affaires de l'établissement941(*). Or l'activité de cession des PSL connaît une baisse tendancielle continue depuis 2012, qui s'explique par deux raisons942(*) :
- un moindre recours des établissements de santé aux concentrés de globules rouges et au plasma thérapeutique, du fait de l'évolution des pratiques médicales qui permettent notamment des chirurgies moins invasives ;
- l'ouverture à la concurrence du marché du plasma thérapeutique depuis un arrêt du Conseil d'État de 2014.
La situation de l'EFS s'est également trouvée fragilisée fin 2019 par la fin du taux de TVA à 2,1 % applicable sur une part d'activité de l'établissement, qui a entraîné une perte de recette alors estimée à plus de 75 millions d'euros943(*).
Dans son rapport public annuel 2019 précité, la Cour des comptes jugeait la filière sang « menacée » du fait de son modèle économique et appelait à une réaction rapide des pouvoirs publics.
Pour permettre à l'EFS de faire face à cette situation, il a été décidé de lui accorder un soutien transitoire, sous la forme d'une dotation dégressive de l'assurance maladie qui devait s'éteindre en 2023. Un plan de transformation devait être conduit par l'EFS au cours de la période 2019-2022, en contrepartie de ce soutien financier, pour contribuer à restaurer durablement sa situation financière. Toutefois, celui-ci n'a pas suffi à équilibrer le budget de l'EFS, la crise sanitaire de la covid-19 ayant notamment accentué la baisse d'activité subie par l'EFS sur la cession des PSL.
Le tableau ci-après retrace le montant annuel des dotations accordées depuis 2019. Si les dotations sont bien dégressives entre 2020 et 2022, l'EFS ayant clôturé l'année 2022 avec un déficit de près de 40 millions d'euros, une nouvelle dotation de 55 millions d'euros versée par l'assurance maladie a donc été accordée.
Les dotations accordées par la branche maladie à l'EFS depuis 2019
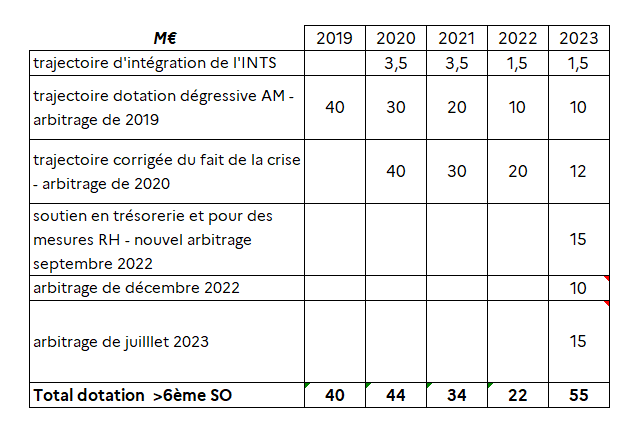
Source : Réponse du Gouvernement à une question posée dans la perspective de l'examen du présent PLFSS sur la base des articles L.O. 111-8 et L.O. 111-9 du code de la sécurité sociale
En outre, une hausse de tarifs des PSL a permis d'augmenter de 20 millions d'euros944(*) les recettes en 2023. Au global, l'apport financier à l'EFS pour 2023 a donc été porté à 75 millions d'euros.
La situation économique de l'EFS, telle que décrite par un récent rapport d'information de la Mecss de la commission des affaires sociales du Sénat
« L'EFS est confronté à un effet ciseau qui met en péril son équilibre financier.
Auditionné, l'EFS, dont les régimes obligatoires de base ne sont qu'un financeur mineur, estime que son modèle financier, fondé principalement sur les produits de la cession de produits sanguins labiles aux établissements de santé et au laboratoire français du fractionnement et des biotechnologies, arrive à épuisement. En effet, la cession de concentrés de globules rouges, qui représente les deux tiers des produits de l'établissement, présente une tendance baissière et difficilement prévisible depuis 2012. Cette tendance a été accrue pendant la crise sanitaire, avec une fréquentation amoindrie des collectes et avec une activité hospitalière hors covid-19 en retrait.
Dans sa contribution écrite à la mission, l'EFS note que « depuis plusieurs années, le modèle économique de l'EFS est marqué par des tensions croissantes sur ses équilibres financiers. L'établissement apparaît sous-financé. » L'EFS a ainsi exécuté des budgets en déficit de 11 millions d'euros en 2019, puis de 40 millions d'euros en 2022.
Cette baisse des recettes se conjugue, depuis 2022, avec une augmentation des dépenses. L'établissement décrit ainsi un double choc pour les finances de l'EFS : un choc inflationniste de 30 millions d'euros et un choc d'attractivité responsable d'une hausse de près de 35 millions d'euros des dépenses de personnel afin de répercuter le Ségur de la santé sur les salaires des effectifs de l'établissement.
Dans ce contexte, l'attribution d'une dotation pérenne de l'assurance maladie peut être une piste à développer.
Il est à noter, à ce titre, qu'une mission conjointe de l'inspection générale des finances et de l'inspection générale des affaires sociales réalise, sur demande de quatre ministres, une mission sur le modèle économique de l'EFS, qui devrait rendre ses conclusions courant 2023. »
Source : Élisabeth Doineau, Annie Le Houerou, Dotations de la sécurité sociale : sortir de la logique du financement à l'aveugle, rapport d'information n° 877 (2022-2023), mission d'évaluation et de contrôle de la sécurité sociale (Mecss) de la commission des affaires sociales du Sénat, 12 juillet 2023.
B. La réforme du financement de l'EFS
1. La prise en compte de coûts de revient dans la réglementation des tarifs des PSL
Le nouveau modèle s'appuiera toujours à titre principal sur les recettes issues de la cession des PSL, qui doivent constituer environ 85 % du total des recettes, soit 850 millions d'euros. En revanche, les tarifs réglementés des PSL prendront davantage en compte le coût de revient réel des PSL, c'est-à-dire leur coût de production.
Ces coûts de revient devront progressivement intégrer l'impact de l'amélioration de la performance de l'établissement permise par la conduite d'un plan de transformation et d'efficience, qui constituera une feuille de route de la nouvelle présidence de l'EFS945(*). Il pourrait s'agir notamment d'intervenir en priorité sur la rationalisation des fonctions supports, les gains logistiques et de transport et un meilleur dimensionnement des activités de collectes.
En conséquence, le I du présent article tend à compléter l'article L. 164-1 du code de la sécurité sociale946(*) par un alinéa prévoyant que « le tarif de cession des produits [des activités liées aux produits sanguins labiles947(*)] cédés pour une finalité transfusionnelle est déterminé en tenant compte du coût de revient de la collecte, la qualification biologique, la préparation, la distribution, la délivrance et le contrôle de la qualité desdits produits incombant à l'Établissement français du sang ».
2. La pérennisation d'une dotation de l'assurance maladie pour contribuer au financement de missions de service public
Le nouveau modèle de financement de l'EFS aura un caractère « mixte » puisque les recettes issues de la cession des PSL seront complétées par une dotation pérenne de l'assurance maladie. L'étude d'impact du Gouvernement indique que son montant devrait s'établir à 100 millions d'euros, soit environ 10 % des recettes de l'EFS.
Le II du présent article propose de préciser les conditions de versement de cette dotation. Pour cela, il prévoit de remplacer les 3° et 4° de l'article L. 1222-8 du code de la santé publique par deux nouveaux alinéas. La rédaction actuelle prévoit :
« 3° Des redevances pour services rendus établies par décret dans les conditions fixées par l' article 5 de l'ordonnance n° 59-2 du 2 janvier 1959 portant loi organique relative aux lois de finances ;
4° Des produits divers, des dons et legs ainsi que des subventions de l'État, des collectivités publiques, de leurs établissements publics et des organismes d'assurance maladie ; La participation des organismes d'assurance maladie est versée et répartie entre les régimes dans des conditions fixées par décret ; »
Dans la rédaction proposée, le 3° vise désormais une dotation des régimes obligatoires d'assurance maladie, dont le montant est fixé chaque année par arrêté ministériel. Cette dotation contribue au financement à la fois de missions de service public et de surcoûts temporaires non couverts par les tarifs réglementés des PSL.
L'annexe 9 au présent PFLSS948(*) précise que parmi les missions de service public, est visé en particulier l'accès aux produits sanguins sur l'ensemble du territoire métropolitain et ultramarin dans les délais compatibles avec les besoins hospitaliers, soit une activité sept jours sur sept et 24 heures sur 24949(*).
S'agissant des impacts financiers non couverts par le mécanisme tarifaire, l'annexe 9 indique que sont notamment visés les investissements dédiés au renouvellement des actifs, et plus particulièrement les investissements nécessaires au développement de la collecte de plasma, ainsi que les coûts liés à la conduite des chantiers de modernisation.
Il est également indiqué que « la participation des organismes d'assurance maladie est versée et répartie entre les régimes ». Cette rédaction est issue de la disposition actuelle du 4° de l'article L. 1222-8 précité, selon laquelle « la participation des organismes d'assurance maladie est versée et répartie entre les régimes dans des conditions fixées par décret »950(*).
Quant à la nouvelle rédaction du 4°, elle ajuste à la marge la rédaction actuelle en supprimant la référence aux organismes d'assurance maladie, désormais prévue au 3°.
Au final, une dotation de 100 millions d'euros versée à l'EFS par l'assurance maladie représente une hausse de 45 millions d'euros par rapport aux 55 millions d'euros versés en 2023 par l'assurance maladie.
II - Les modifications considérées comme adoptées par l'Assemblée nationale
Dans le texte sur lequel il a engagé sa responsabilité devant l'Assemblée nationale en application de l'article 49 alinéa 3 de la Constitution, le Gouvernement a retenu un amendement rédactionnel.
Cet article est considéré comme ayant été adopté par l'Assemblée nationale, ainsi modifié.
III - La position de la commission
La commission se félicite qu'une mesure qui assure la pérennité et la viabilité du modèle de financement de l'EFS figure dans ce projet de loi de financement de la sécurité sociale. Cette mesure s'inscrit d'ailleurs dans la continuité d'un rapport d'information951(*) de la mission d'évaluation et de contrôle de la sécurité sociale (Mecss) sur les organismes et fonds financés par les régimes obligatoires de base (Offrob), qui soulignait notamment l'importance de promouvoir un modèle de financement transparent des Offrob, parmi lesquels figure l'EFS, dans une logique de pluriannualité et de transparence pour renforcer l'information et l'exercice du contrôle parlementaires.
Si la sécurisation des ressources de l'EFS ne doit pas attendre que l'ensemble des réformes nécessaires à l'amélioration de la performance de l'établissement soient mises en oeuvre et produisent tous leurs effets, la commission souligne la nécessité d'en suivre la mise en oeuvre effective chaque année. En particulier, il serait regrettable qu'un échec relatif de la politique d'efficience conduise à employer la dotation de l'assurance maladie pour financer d'autres activités que celles précisément identifiées, par exemple l'activité de collecte, qui a d'abord vocation à être financée par les tarifs réglementés.
En parallèle, la commission attire l'attention du Gouvernement sur des enjeux qui ne lui semblent pas suffisamment identifiés :
- le risque de réduction des capacités ou de dégradation de l'outil de collecte, compte tenu de la fragilité de la participation des donneurs et de la difficulté à prévoir les besoins, exige une vigilance particulière pour éviter de se trouver confrontés à un besoin de produits sanguins non anticipé, par exemple en situation de crise, sans être en mesure d'y répondre ;
- la nécessité de disposer d'un outil de production adapté à la législation européenne et permettant de se positionner durablement sur le marché concurrentiel du plasma thérapeutique. Or, comme cela a été indiqué à la commission, les retards d'investissement s'accumulent, qu'il s'agisse du parc immobilier, des équipements matériels tels que les structures de prélèvement ou des équipements informatiques ;
- le soutien de l'attractivité de l'EFS en matière de ressources humaines, dès lors que la structure souffre d'un turnover important et d'une difficulté à fidéliser les personnels en raison d'écarts de rémunérations qui se creusent avec les établissements de santé. La commission souligne la nécessité d'engager un plan de revalorisation sans tarder.
La réforme de l'EFS, et plus généralement de la filière sang, est un enjeu stratégique majeur de santé publique et de souveraineté industrielle, qui suppose un positionnement proactif dans la production des thérapies innovantes. En conséquence, la commission souhaite que le Parlement soit étroitement associé aux décisions relatives à l'évolution des montants des dotations qui seront allouées à l'EFS à l'avenir.
Enfin, la commission a adopté un amendement n° 287 tendant à préciser, dans la rédaction proposée pour l'article L. 164-1 du code de la santé publique, que les tarifs dont il est question sont seulement les tarifs de l'EFS fixés par arrêté ministériel, et non ceux du plasma thérapeutique, qui sont libres, ce plasma étant aussi commercialisé par des entreprises pharmaceutiques privées.
Elle a également adopté un amendement n° 288 tendant à préciser la rédaction proposée pour l'article L. 1222-8 du code de la santé publique.
En conséquence, la commission propose d'adopter cet article, sous réserve de ces précisions rédactionnelles.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article
32
Utilisation des préparations officinales spéciales dans le
cadre du plan blanc
Cet article vise à étendre le statut des préparations hospitalières spéciales aux cas d'arrêt de commercialisation d'un médicament d'intérêt thérapeutique majeur et à permettre leur dispensation en officine. Il crée, par ailleurs, le statut des préparations officinales spéciales, destinées à faire face à une rupture de stock, un arrêt de commercialisation ou une crise sanitaire.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - Le dispositif proposé
A. Les pharmacies ont été progressivement impliquées ces dernières années pour restaurer l'offre de médicaments en rupture
1. Multifactorielles, les pénuries de médicaments peuvent survenir même en l'absence de tension sur les principes actifs
Les pénuries et tensions d'approvisionnement en médicaments, plus précisément analysées infra952(*), constituent un phénomène complexe, mondial et en constante aggravation, alimenté par de nombreux facteurs structurels analysés récemment par une commission d'enquête sénatoriale953(*). Parmi eux, peuvent notamment être cités :
- la rapide augmentation de la demande mondiale, progressivement solvabilisée par la mise en place et le renforcement des systèmes de protection sociale nationaux954(*) ;
- la fragmentation industrielle et géographique du secteur, portée par le recours croissant des laboratoires à la sous-traitance, singulièrement pour la production de produits matures peu rentables955(*) ;
- la production en flux tendu, rendant l'approvisionnement sensible à des incidents industriels pourtant fréquents956(*) ;
- les choix stratégiques de certains laboratoires, privilégiant les produits innovants et onéreux au détriment de produits matures moins avantageusement tarifés957(*).
En conséquence, les causes concrètes des pénuries apparaissent variables et indépendantes, souvent, de l'approvisionnement en principes actifs.
Les industriels étant contraints, depuis 2016958(*), de lui déclarer toute rupture ou risque de rupture de stock et, depuis 2019959(*), de le faire dès qu'ils en ont connaissance960(*), l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) dispose désormais de données précises sur le nombre et les causes déclarées des pénuries constatées, qu'elle publie annuellement.
En 2021 comme en 2022, moins de 8 % des ruptures et risques de rupture recensés trouvaient leur cause, d'après les déclarations reçues par l'ANSM, dans un défaut d'approvisionnement en matière première.
Répartition des causes déclarées des ruptures et risques de ruptures en 2021 et 2022

Source : Commission des affaires sociales du Sénat, d'après des données publiées par l'ANSM
Plus souvent, l'augmentation subite de la demande, ou les capacités insuffisantes de production - y compris en matière de formulation ou de conditionnement de médicaments pour lesquels la matière première pharmaceutique demeure disponible -, sont mises en avant.
Dans de telles situations, les pharmacies ont été amenées, ces dernières années, à contribuer à l'effort de maintien de la disponibilité de médicaments en rupture particulièrement indispensables à la prise en charge des patients.
2. Les pénuries de curares, survenues durant la crise sanitaire, ont conduit à la reconnaissance des préparations hospitalières spéciales
Les services de réanimation hospitaliers ont connu, durant la crise sanitaire, de graves tensions d'approvisionnement pour cinq spécialités pourtant indispensables à la prise en charge des patients : le cisatracurium, l'atracurium, le rocuronium, la kétamine et le midazolam, utilisés notamment pour l'anesthésie.
En réponse à cette situation, provoquée par l'augmentation subite et importante de la demande en lien avec l'aggravation de l'épidémie de covid-19, un dispositif de régulation nationale a été mis en place, mobilisant notamment l'établissement pharmaceutique (EP) de l'Assistance publique-Hôpitaux de Paris (AP-HP) et un réseau de six pharmacies à usage intérieur (PUI) hospitalières961(*).
À la demande de la Direction générale de la santé (DGS), un partenariat a été mis en place entre l'EP de l'AP-HP, également appelé Agence générale des équipements et produits de santé (Ageps), un EP privé, l'ANSM et les centres hospitaliers de Lille et de Lyon pour la production de préparations hospitalières injectables de cisatracurium.
Ce partenariat a, d'après le Gouvernement, permis la mise à disposition d'environ 200 000 ampoules produites, sur la base d'une monographie établie par le réseau des PUI impliquées puis validée par l'ANSM et grâce à la fourniture du principe actif par un laboratoire privé, par les PUI de Lille et de Lyon, sous la coordination de l'Ageps962(*).
Pour tirer les conséquences de cette expérience et permettre sa reproduction dans les cas de pénurie les plus graves, la LFSS pour 2022 a créé la catégorie des préparations hospitalières spéciales (PHS)963(*).
Du fait des difficultés techniques de leur fabrication ou de la faible disponibilité des substances actives nécessaires, celles-ci ont vocation à être réalisées dans des PUI, des EP d'établissements de santé ou de Santé publique France, dans des conditions définies par décret en Conseil d'État, ou sous leur responsabilité lorsqu'ils confient à titre exceptionnel la réalisation des préparations à un EP privé964(*).
Les PHS doivent faire l'objet d'une autorisation précisant leurs modalités de réalisation, délivrée :
- par le directeur général de l'ANSM, dans les cas de rupture de stock d'un médicament d'intérêt thérapeutique majeur (MITM) ;
- ou par le ministre de la santé, pour faire face à une menace ou à une crise sanitaire grave965(*).
Attendu depuis et plusieurs fois réclamé par le Sénat966(*), le décret en Conseil d'État nécessaire à l'application de ces dispositions n'a, toutefois, toujours pas été publié.
3. Les pharmacies d'officine ont, durant l'hiver 2022-2023, été mobilisées dans la préparation de spécialités en rupture
Les préparations des pharmacies d'officine sont, aujourd'hui, strictement encadrées par le code de la santé publique.
Les préparations officinales y sont définies comme tout médicament préparé en pharmacie, inscrit à la pharmacopée ou au formulaire national et destiné à être dispensé directement aux patients approvisionnés par cette pharmacie. Elles se distinguent des préparations magistrales en ce qu'elles ne sont pas nécessairement exécutées selon une prescription médicale et destinées à un malade déterminé967(*).
L'exécution des préparations officinales et magistrales figure parmi les missions des pharmacies d'officine. Celles-ci peuvent confier l'exécution d'une préparation, par un contrat écrit, à une autre officine qui est alors soumise, pour l'exercice de cette activité de sous-traitance, à une autorisation préalable délivrée par le directeur général de l'ARS968(*).
Les préparations sont soumises à plusieurs contraintes de sécurité et de qualité. Elles doivent être, d'abord, exécutées en conformité avec les bonnes pratiques définies par décision de l'ANSM969(*). Les officines sont, par ailleurs, tenues de disposer, dans la partie non accessible au public, d'un local réservé à l'exécution et au contrôle des préparations magistrales ou officinales970(*). Le directeur général de l'ANSM peut suspendre ou interdire l'exécution des préparations, lorsque l'officine ne respecte pas les bonnes pratiques de préparation ou réalise les préparations dans des conditions dangereuses pour la santé publique971(*).
Enfin, les préparations pouvant présenter un risque pour la santé sont soumises à un régime d'autorisation préalable972(*). Un arrêté de 2014 classe dans cette catégorie les préparations stériles, les préparations à partir de produits cancérogènes, mutagènes ou toxiques pour la reproduction, ainsi que la plupart des préparations destinées aux enfants de moins de douze ans973(*).
Malgré ce cadre restrictif, les préparations des pharmacies d'officine se sont avérées utiles, durant l'hiver 2022-2023, pour maintenir la disponibilité de médicaments en rupture pourtant essentiels à la prise en charge des patients.
Pour permettre leur implication, l'ANSM a, dès décembre 2022, publié les monographies des préparations magistrales d'amoxicilline 125 mg et 250 mg et autorisé974(*) les pharmaciens à délivrer une préparation magistrale d'amoxicilline lorsque le médicament prescrit n'était pas disponible975(*).
Une quarantaine de pharmacies ont ainsi été autorisées à exécuter ces préparations et permis de répondre, notamment, aux demandes de dosages spécifiques dans le cadre de prises en charge pédiatriques976(*).
B. L'article 32 vise à étendre les cas de recours aux PHS et crée le statut des préparations officinales spéciales (POS)
L'article 32 vise à renforcer le statut des PHS et à consacrer, dans le code de la santé publique, le statut des préparations officinales spéciales (POS) pouvant être réalisées par les pharmacies d'officine.
Pour ce faire, le 1° du I apporte deux modifications à l'article L. 5121-1 du code de la santé publique définissant les préparations hospitalières spéciales, destinées à renforcer ce statut.
D'une part, il élargit les cas dans lesquels le directeur général de l'ANSM peut autoriser, à titre exceptionnel et temporaire, la réalisation de préparations hospitalières spéciales. Cette autorisation pourrait non plus seulement faire suite à une rupture de stock d'un MITM, mais également à un arrêt de commercialisation.
D'autre part, il permet au ministre de la santé d'autoriser, à titre dérogatoire et afin de répondre à l'ensemble des besoins nationaux, la dispensation des PHS par les pharmacies d'officine.
Le 2° du I complète les mêmes dispositions pour créer le statut des POS. Pour faire face à une rupture de stock ou un arrêt de commercialisation d'un MITM, à une menace ou à une crise sanitaire grave et pour garantir la qualité et la sécurité d'utilisation des produits, le ministre de la santé pourra autoriser par arrêté, à titre exceptionnel et temporaire, la réalisation de POS.
La réalisation de POS sera réservée aux officines disposant d'une autorisation du directeur général de l'ARS pour exécuter des préparations pouvant présenter un risque pour la santé. Les POS seront, par ailleurs, soumises à trois exigences :
- être soumises à prescription médicale ;
- être réalisées selon une monographie publiée par l'ANSM ;
- être préparées à partir d'une matière première à usage pharmaceutique fournie par l'établissement pharmaceutique d'un établissement de santé, soit par l'Ageps.
Un décret en Conseil d'État devra définir les conditions d'application de ces dispositions.
Le II de l'article 32 insère au sein du code de la sécurité sociale un nouvel article L. 162-16-4-5 prévoyant que sont fixés par arrêté conjoint des ministres de la santé et de la sécurité sociale :
- les prix de cession, couvrant les frais de réalisation et les frais de dispensation en officine, des PHS faisant l'objet d'une dispensation en officine ;
- les prix de cession des POS.
II - Les modifications adoptées par l'Assemblée nationale
Le Gouvernement n'a retenu, dans le texte sur lequel il a engagé sa responsabilité devant l'Assemblée nationale en application du troisième alinéa de l'article 49 de la Constitution, que trois amendements rédactionnels de la rapporteure générale.
Cet article est considéré comme ayant été adopté par l'Assemblée nationale, ainsi modifié.
III - La position de la commission
La commission a soutenu, lors de l'examen du PLFSS pour 2022, la création du statut des PHS et souhaité renforcer le rôle des pharmacies d'officine dans ces dispositions977(*).
Elle souligne, à cet égard, que la contribution de l'Ageps et des PUI hospitalières au maintien de la disponibilité de médicaments à fort enjeu thérapeutique, dans les situations de pénurie ou de risque de pénurie les plus graves, constitue un enjeu essentiel.
Le statut des PHS apparaît, de ce point de vue, indispensable. Comme l'a souligné l'AP-HP, interrogée par la rapporteure, celui-ci permet de mettre à disposition des patients des préparations bien plus rapidement que ne le permettrait une nouvelle demande d'autorisation de mise sur le marché (AMM), impliquant un délai d'attente supérieur à un an978(*). Pour la même raison, le statut des PHS est susceptible de s'avérer utile en cas d'arrêt de commercialisation et dans l'attente de la délivrance d'une nouvelle AMM à un éventuel repreneur.
La commission a, en conséquence, soutenu les dispositions du présent article visant à permettre le recours aux PHS dans les cas d'arrêt de commercialisation et la dispensation de celles-ci dans les pharmacies d'officine.
Elle s'est étonnée, en revanche, de trouver dans le présent texte l'extension d'un cadre juridique dont le Gouvernement n'a toujours pas permis, deux ans après son adoption, la mise en oeuvre effective. Compte tenu des objectifs attachés à ce nouveau statut comme du caractère largement imprévisible des tensions d'approvisionnement en médicaments, la commission juge difficilement explicable le retard accumulé dans la publication du décret en Conseil d'État attendu.
Elle rappelle que l'ancien ministre de la santé et de la prévention, plusieurs fois alerté à ce sujet à l'occasion de travaux du Sénat, a déclaré lors d'une audition de la commission d'enquête sénatoriale relative aux pénuries de médicaments que sa publication devrait intervenir en octobre 2023, après transmission à la Commission européenne979(*).
La commission appelle le Gouvernement à permettre sans plus attendre l'application de ce nouveau statut.
Soucieuse de permettre l'implication des pharmacies d'officine à l'effort de lutte contre les pénuries de médicaments, et consciente de leurs compétences en matière de préparation, la commission avait adopté un amendement au PLFSS pour 2022 ouvrant la possibilité aux PUI et EP publics mobilisés dans la production de PHS d'en confier l'exécution, partiellement ou entièrement, à des pharmacies d'officine sous-traitantes dûment autorisées980(*). Celui-ci n'avait, finalement, pas été retenu dans le texte définitivement adopté par l'Assemblée nationale.
La commission, qui demeure attachée à une plus grande implication des pharmacies d'officine dans la production de médicaments en rupture, a adopté à nouveau, à l'initiative de sa rapporteure, un amendement n° 289 portant la même modification.
Pour les mêmes raisons, la commission a favorablement accueilli les dispositions du présent article créant le statut des POS. Elle souligne que, tenant compte notamment du rôle joué par les officines face aux difficultés d'approvisionnement en amoxicilline survenues durant l'hiver 2022-2023, la commission d'enquête sénatoriale sur les pénuries de médicaments avait recommandé la création d'un tel statut981(*).
Consciente de la compétence de l'Ageps en matière de contrôle et de qualification de la matière première à usage pharmaceutique (MPUP), la commission a toutefois jugé inopportun de limiter, dans la loi, l'approvisionnement des officines aux seules MPUP fournies par elle. Une telle obligation, en situation d'urgence, pourrait ralentir la mise en oeuvre des POS, alors que leur objet est précisément d'offrir un nouvel outil susceptible de permettre de répondre, dans l'urgence, aux situations de pénurie sur des médicaments à fort enjeu thérapeutique.
La commission rappelle, à cet égard, que les activités de fabrication, d'importation et de distribution de substances actives sont strictement encadrées par le code de la santé publique.
Elles ne peuvent être exercées que dans des établissements autorisés par l'ANSM982(*). L'importation de substances actives n'est, de plus, permise que lorsque celles-ci ont été fabriquées conformément à des normes de bonnes pratiques au moins équivalentes983(*).
Aussi la commission a-t-elle adopté un amendement n° 290 de sa rapporteure permettant l'exécution de POS à partir d'une matière première fournie, dans les conditions prévues par le décret en Conseil d'État attendu, par tout établissement autorisé par l'ANSM.
La commission a également souhaité mieux associer les organisations représentatives des pharmaciens d'officine à la définition des prix de cession des POS et, lorsqu'elles font l'objet d'une dispensation en officine, des PHS. Elle a, en conséquence, adopté un amendement n° 291 de sa rapporteure prévoyant que l'arrêté fixant ces tarifs doit être pris après leur consultation.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article
33
Renforcement des leviers d'épargne de médicaments en cas de
rupture d'approvisionnement
Cet article vise à élever au niveau législatif la définition des ruptures d'approvisionnement et, dans de telles situations, à maîtriser le niveau de prescription et de dispensation en permettant au Gouvernement de rendre obligatoire le recours aux ordonnances conditionnelles ou la délivrance de médicaments à l'unité, ainsi que de limiter ou interdire la prescription par un acte de télémédecine.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - Le dispositif proposé
A. Les phénomènes de pénurie de médicament se sont aggravés ces dernières années malgré la mise en place de nombreux dispositifs légaux visant à les juguler
1. L'aggravation des phénomènes de pénurie
Face à l'aggravation des difficultés d'approvisionnement, les pouvoirs publics ont cherché ces dernières années à mieux appréhender les phénomènes de pénurie de médicaments.
Définies dans le code de la santé publique depuis l'intervention d'un décret de septembre 2012984(*), les ruptures d'approvisionnement correspondent à l'incapacité, pour une pharmacie d'officine ou une pharmacie à usage intérieur (PUI) hospitalière de dispenser un médicament à un patient dans un délai de 72 heures, après avoir effectué une demande d'approvisionnement auprès de deux entreprises exerçant une activité de distribution de médicaments.
Ces ruptures d'approvisionnement peuvent être imputables à une rupture de stock, laquelle est définie comme l'impossibilité de fabriquer ou d'exploiter un médicament985(*).
Les obligations déclaratives des exploitants ont, également, progressivement été renforcées. La loi de modernisation de notre système de santé de 2016986(*) a, ainsi, fait obligation aux industriels de déclarer à l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) tout risque de rupture et toute rupture de stock sur les médicaments qu'ils exploitent. La loi relative à l'organisation et à la transformation du système de santé (OTSS) de 2019987(*) a renforcé cette obligation en précisant que les exploitants doivent procéder à cette déclaration dès qu'ils ont connaissance de tels risques988(*).
Ces déclarations permettent à l'ANSM de recenser les difficultés d'approvisionnement et de retracer, chaque année, leur évolution. À cet égard, le nombre de ruptures de stock ou de risques de rupture déclarés par les exploitants apparaît avoir fortement augmenté ces dernières années989(*).
Nombre de ruptures de stock et de risques de rupture déclarés à l'ANSM par les exploitants (2014-2022)
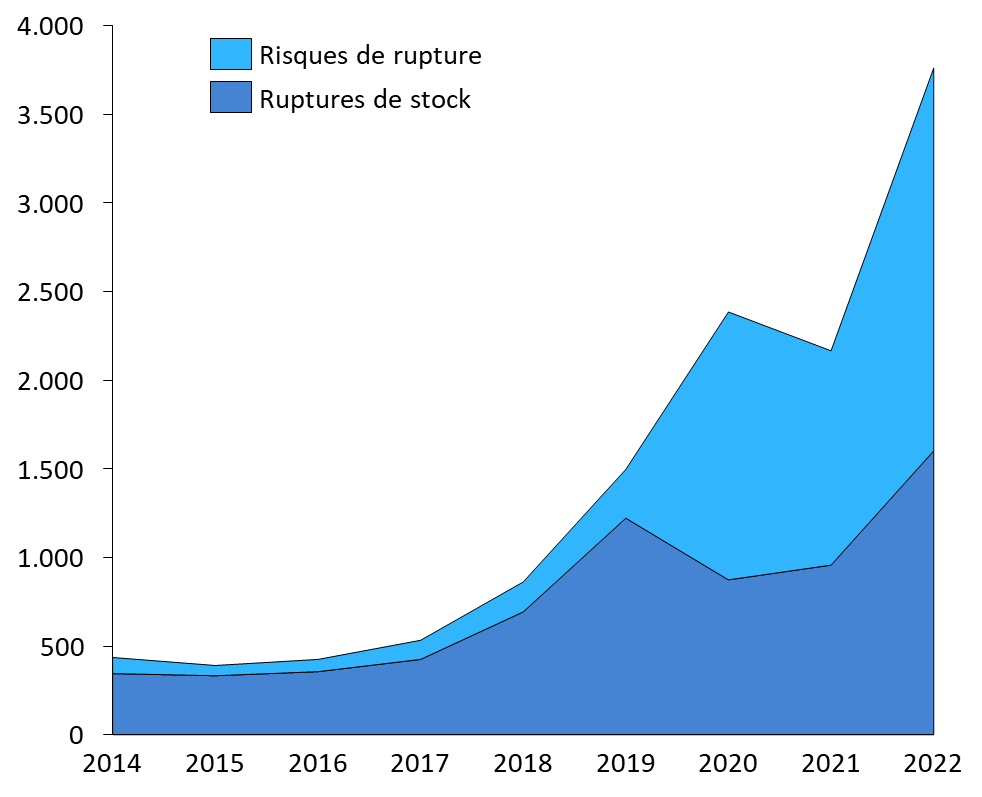
Source : Commission des affaires sociales du Sénat, d'après des données publiées par l'ANSM
Les ruptures et risques de rupture recensés touchent l'ensemble des quatorze classes thérapeutiques distinguées par la nomenclature anatomique, thérapeutique et chimique (dite « classification ATC »). D'après l'ANSM, concentrent toutefois le plus fort nombre de difficultés déclarées les médicaments :
- du système cardio-vasculaire (1 087 déclarations en 2022) ;
- du système nerveux (721 déclarations en 2022) ;
- anti-infectieux à usage systémique (554 déclarations en 2022)990(*).
Les difficultés d'approvisionnement concernent, enfin, majoritairement des produits matures. Selon la commission d'enquête sénatoriale relative à la pénurie de médicaments et aux choix de l'industrie pharmaceutique française, entre 60 % et 70 % des déclarations de rupture visent des médicaments dont l'autorisation de mise sur le marché (AMM) a été octroyée il y a plus de dix ans991(*).
2. Le renforcement des obligations des exploitants et distributeurs
Pour juguler les phénomènes de pénurie, les contraintes légales et réglementaires imposées aux exploitants et aux distributeurs ont progressivement été renforcées. Pour l'essentiel, celles-ci sont concentrées sur les médicaments d'intérêt thérapeutique majeur (MITM), que la loi « santé » de 2016992(*) a définis comme étant ceux pour lesquels une interruption de traitement :
- est susceptible de mettre en jeu le pronostic vital des patients à court ou moyen terme ;
- ou représente une perte de chance importante pour les patients au regard de la gravité ou du potentiel évolutif de la maladie993(*).
Les exploitants ont, ainsi, été responsabilisés de manière croissante dans l'anticipation et la gestion des ruptures.
En matière d'anticipation, les industriels sont, depuis la loi de financement de la sécurité sociale (LFSS) pour 2020994(*), tenus d'établir un plan de gestion des pénuries (PGP) pour chacun des MITM qu'ils exploitent995(*). Ces derniers doivent, notamment, tenir compte des risques relatifs au cycle de fabrication et de distribution de la spécialité concernée et identifier les médicaments susceptibles de constituer une alternative thérapeutique. Ils peuvent prévoir d'autres sites de fabrication de matières premières, de formulation ou de conditionnement996(*).
En matière de détection des pénuries, les industriels doivent notamment, lors de leurs déclarations obligatoires de toute rupture ou de tout risque de rupture, préciser les stocks disponibles, les délais prévisionnels de remise à disposition ainsi que, le cas échéant, les spécialités substituables997(*).
Enfin, les industriels sont également appelés à permettre une meilleure gestion des périodes de tension. Ils sont tenus de disposer de centres d'appel d'urgence permanents accessibles aux pharmaciens998(*) et de mettre en oeuvre, en situation de rupture et en lien avec l'ANSM, les mesures prévues par les PGP transmis999(*).
Surtout, la LFSS pour 20201000(*) a contraint les industriels à constituer un stock de sécurité destiné au marché national, destiné à être exploité dans les périodes de tension pour laisser aux acteurs le temps de mettre en oeuvre les mesures de résolution appropriées. Ces stocks doivent correspondre à au moins deux mois de couverture des besoins pour les MITM et une semaine pour les médicaments ne relevant pas de cette catégorie1001(*).
Les contraintes des grossistes-répartiteurs ont également été renforcées.
La loi « Médicaments » de 20111002(*), précisée par un décret de septembre 20121003(*), leur a ainsi imposé de respecter les obligations de service public suivantes :
- disposer d'un assortiment de médicaments comportant au moins neuf dixièmes des présentations commercialisées en France ;
- être en mesure de satisfaire la consommation de sa clientèle habituelle durant au moins deux semaines et à tout moment, à l'exception des samedis après 14 heures, dimanches et jours fériés ;
- livrer toute commande dans les vingt-quatre heures au sein de leurs territoires respectifs de répartition1004(*).
Depuis 2016, la loi interdit, enfin, aux grossistes-répartiteurs d'exporter des MITM pour lesquels une rupture ou un risque de rupture a été mis en évidence et qui figurent, en conséquence, sur une liste établie par l'ANSM1005(*). Ils ne sont, par ailleurs, autorisés à exporter les autres médicaments que dans la mesure où ils ont rempli leurs obligations de service public1006(*).
La commission d'enquête sénatoriale précitée a toutefois souligné que ces obligations demeuraient insuffisamment appliquées. La qualité des PGP transmis est, selon l'ANSM elle-même et d'après les contrôles réalisés par la commission d'enquête, fortement inégale1007(*).
Si l'ANSM dispose d'un pouvoir de sanction financière introduit par la loi « Médicaments » et progressivement renforcé depuis, susceptible de s'appliquer aux exploitants ne respectant pas les obligations prévues1008(*), ces sanctions demeurent pour le moment peu utilisées et n'ont concerné entre 2018 et 2022 que des manquements à l'obligation de déclaration d'une rupture ou d'un risque de rupture1009(*).
3. Les mesures prises par l'ANSM et les efforts de maîtrise des prescriptions et des dispensations
Indépendamment des obligations imposées aux exploitants et distributeurs, les pouvoirs publics ont pris des mesures ces dernières années visant à mieux gérer ou prévenir les phénomènes de pénurie.
Plusieurs évolutions législatives et réglementaires ont d'abord visé à mieux maîtriser le volume des prescriptions ou améliorer la gestion des pénuries.
Pour maîtriser les prescriptions d'antibiotiques, encore importantes en France, la réalisation de tests rapides d'orientation diagnostique (Trod) par les pharmaciens et d'autres professionnels de santé a, ainsi, été permise par un arrêté de 20161010(*) et progressivement encouragée. La LFSS pour 2020 a, par ailleurs, créé le dispositif des ordonnances conditionnelles1011(*), permettant au prescripteur de conditionner la délivrance de certains médicaments à la réalisation de tests et à l'obtention de résultats déterminés1012(*). L'incitation au recours au Trod figure, enfin, parmi les actions prévues par la stratégie nationale 2022-2025 de prévention des infections et de l'antibiorésistance1013(*).
D'autres évolutions ont visé à faciliter la gestion des pénuries. La loi « santé » de 2016 a, ainsi, permis à l'ANSM d'autoriser les pharmacies d'officine à dispenser au détail des médicaments disposant d'une autorisation d'importation de l'ANSM en situation de rupture d'un MITM1014(*). La loi OTSS de 2019 a, par ailleurs, autorisé les pharmaciens, en cas de rupture ou de risque de rupture de stock, à remplacer un MITM prescrit par un autre médicament conformément à une recommandation établie par l'ANSM et publiée sur son site internet1015(*). Enfin, la loi de 2020 relative à la lutte contre le gaspillage et à l'économie circulaire a permis la délivrance de certains médicaments en officine, lorsque leur forme le permet, à l'unité1016(*).
L'ANSM est amenée, par ailleurs, à intervenir directement lorsqu'une rupture ou un risque de rupture lui est signalé sur un MITM, en tenant compte du PGP transmis par l'exploitant.
L'Agence indique ainsi qu'en 2022, 42,5 % des déclarations de rupture ou de risque de rupture reçues ont donné lieu à au moins une mesure de gestion de pénurie1017(*). Plus de 85 % des mesures prises ont consisté en :
- la mise en oeuvre d'un contingentement quantitatif, soit d'une distribution en quantité limitée pour maintenir une livraison continue et équitable des stocks disponibles ;
- la constitution d'un stock de dépannage, permettant de répondre à d'éventuels besoins urgents.
Mesures prises par l'ANSM en 2022 suite à des déclarations de rupture ou de risque de rupture
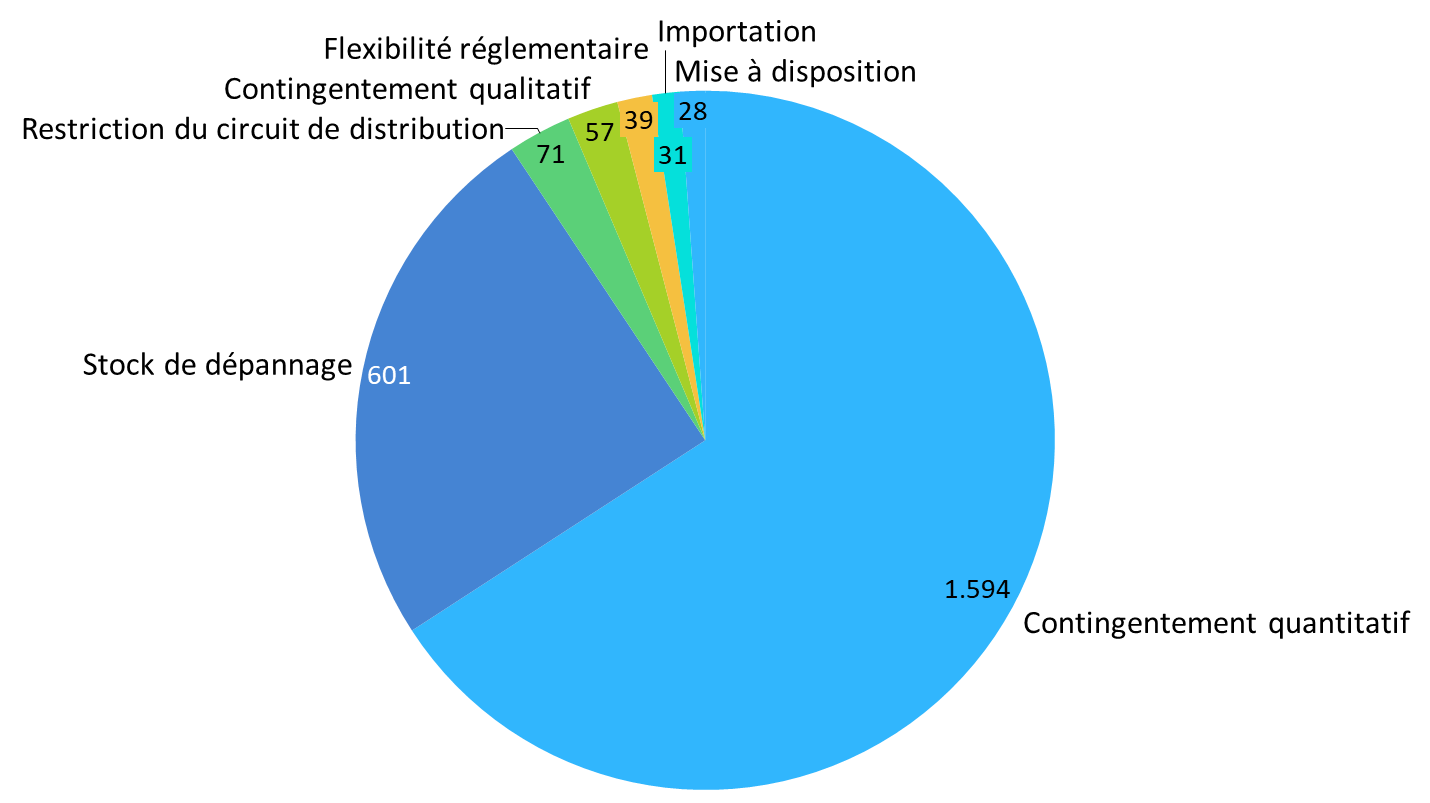
Source : commission des affaires sociales, d'après des données publiées par l'ANSM
B. L'article 33 vise à renforcer, en cas de rupture, les leviers d'épargne de médicaments
Le présent article vise à favoriser la maîtrise des volumes prescrits et dispensés en cas de rupture par la création de trois nouveaux outils légaux susceptibles d'être mobilisés dans une telle situation.
Le 1° modifie l'article L. 5121-29 du code de la santé publique, pour élever au niveau législatif la définition d'une rupture d'approvisionnement. Celle-ci reste attachée à l'incapacité d'une pharmacie d'officine ou d'une PUI de dispenser un médicament dans un délai donné, qui peut être réduit à l'initiative du pharmacien lorsque la poursuite optimale du traitement l'impose. Le délai demeurera défini par décret en Conseil d'État, ainsi que les diligences que le pharmacien doit accomplir pour dispenser le médicament.
Le 2° de l'article 33 insère, dans le texte déposé, deux nouveaux articles dans le code de la santé publique.
Le premier permet au ministre de la santé, en cas de rupture d'approvisionnement, de rendre obligatoire le recours aux ordonnances conditionnelles ou à la délivrance de médicaments à l'unité, dans les conditions d'ores et déjà prévues par les dispositions légales l'autorisant et pour les médicaments que visent ces dispositions. Par arrêté du même ministre, il est mis fin sans délai à ces mesures lorsqu'elles ne sont plus nécessaires.
Le second permet au ministre de la santé, en cas de rupture d'approvisionnement de certains médicaments, d'en limiter ou d'en interdire par arrêté la prescription par un acte de télémédecine. Par arrêté du même ministre, il est mis fin sans délai à ces mesures lorsqu'elles ne sont plus nécessaires.
II - Les modifications adoptées par l'Assemblée nationale
Le Gouvernement a retenu, dans le texte sur lequel il a engagé sa responsabilité devant l'Assemblée nationale en application du troisième alinéa de l'article 49 de la Constitution, trois amendements rédactionnels et un amendement de fond de la rapporteure générale.
Ce dernier insère un nouvel article au sein du code de la santé publique prévoyant qu'en cas de rupture ou de risque de rupture d'approvisionnement d'un MITM ou d'un vaccin, le directeur général de l'ANSM peut, après mise en oeuvre d'une procédure contradictoire, prendre toutes les mesures de police sanitaire nécessaires pour garantir un approvisionnement approprié et continu par les titulaires et exploitants d'une AMM.
Il ajoute, par ailleurs, à l'article L. 5423-9 du code de la santé publique relatif aux pouvoirs de sanction financière de l'ANSM, le fait, pour le titulaire ou l'exploitant de l'AMM d'un MITM ou d'un vaccin, de ne pas mettre en oeuvre les mesures de police sanitaire prises par le directeur général de l'ANSM parmi les manquements pouvant justifier l'application d'une telle sanction.
Cet article est considéré comme ayant été adopté par l'Assemblée nationale, ainsi modifié.
III - La position de la commission
Consciente de la complexité et de la gravité des phénomènes de rupture de médicaments constatés ces dernières années, la commission juge indispensable de donner au Gouvernement et à l'ANSM les moyens d'agir en situation de rupture ou de risque de rupture.
Elle souligne, à cet égard, que la récente commission d'enquête sénatoriale relative aux pénuries de médicament, relevant que l'équipe de l'ANSM affectée à la lutte contre les ruptures ne comptait que 7 équivalents temps plein, a conclu à l'inadéquation de ces moyens avec les missions confiées à l'Agence et à la nécessité de renforcer, en urgence, les moyens humains et matériels dont elle dispose pour contrôler le respect par les industriels de leurs obligations légales et réglementaires1018(*).
Soucieuse de donner, également, à l'ANSM les moyens juridiques d'agir, et de sécuriser les mesures prises par l'Agence dans son activité de lutte contre les ruptures, la commission a favorablement accueilli les dispositions, retenues par le Gouvernement dans le texte sur lequel il a engagé sa responsabilité, visant à consacrer dans la loi les pouvoirs de police sanitaire de l'Agence.
La commission a également soutenu les dispositions soumettant les industriels à des sanctions financières dans le cas où ils ne respecteraient pas les mesures de police sanitaire prises et n'assumeraient pas leur obligation d'approvisionnement approprié et continu. Elle souhaite que ces nouvelles sanctions soient rapidement intégrées aux lignes directrices récemment publiées par l'ANSM et puissent être effectivement appliquées en cas de manquement constaté.
La commission a, toutefois, jugé les trois nouveaux leviers d'épargne de médicaments portés par le présent article inégalement opportuns.
Considérant le développement de l'usage des Trod ces dernières années, elle a jugé crédible la mesure conférant au Gouvernement la faculté de rendre obligatoire l'utilisation des ordonnances conditionnelles pour un médicament en rupture. La Cnam, en effet, fait état d'une multiplication par 6,5 du nombre de Trod réalisés entre 2021 et 20221019(*). Par ailleurs, la réalisation de Trod apparaît désormais dûment valorisée dans la convention pharmaceutique1020(*).
La commission a également jugé souhaitable de permettre au Gouvernement de limiter ou d'interdire, dans certains cas, la prescription de certains médicaments en rupture par téléconsultation. Elle s'est montrée favorable à ce que, dans de telles situations, la prescription de certains antibiotiques doive être justifiée par un examen clinique. Elle a toutefois souhaité que cette mesure ne puisse s'appliquer qu'à des médicaments pour lesquels elle apparaît médicalement justifiée. À cet égard, les exceptions prévues par le Gouvernement dans l'étude d'impact lui sont apparues indispensables. En particulier, une telle mesure ne se justifierait :
- ni pour les prophylaxies antibiotiques qui, prescrites en prévention, ne nécessitent pas d'examen clinique ;
- ni pour les antibiotiques pouvant être prescrits après réalisation d'un Trod par le pharmacien, pour lesquels un recours aux ordonnances conditionnelles, y compris en téléconsultation, devrait être privilégié.
En revanche, la commission s'est interrogée sur le troisième levier proposé, permettant au Gouvernement de rendre obligatoire la dispensation à l'unité de spécialités en rupture. Elle a observé, d'abord, qu'une telle mesure n'aurait que peu d'effet sur la disponibilité des médicaments concernés. La dispensation à l'unité n'apparaît, en effet, utile :
- ni en cas de traitement chronique, pour lesquels la dispensation sera récurrente quel que soit le nombre de médicaments délivrés ;
- ni pour les formes galéniques concentrant le plus grand nombre de ruptures, soit les formes pédiatriques et injectables.
Au demeurant, et compte tenu des fortes contraintes qu'une telle mesure imposerait aux pharmaciens d'officine, la commission a jugé surprenant que l'étude d'impact souligne qu' « il n'est pas attendu que l'acte associé avec la dispensation à l'unité engendre de coût supplémentaire », du fait du plafond applicable et de l'atteinte de ce dernier par les seules dispensations à l'unité obligatoires des stupéfiants.
La commission souligne que les pharmaciens sont, d'ores et déjà, pleinement impliqués dans la lutte contre les pénuries de médicaments et y consacrent en moyenne, d'après la commission d'enquête sénatoriale, une heure par jour1021(*). L'obligation de dispensation à l'unité de certains médicaments s'avérerait nécessairement chronophage pour les pharmacies d'officine et devrait, en conséquence, être dûment rémunérée dans des conditions définies par les partenaires conventionnels.
Compte tenu de l'ensemble de ces doutes, la commission a adopté un amendement n° 292 de sa rapporteure supprimant du présent article la possibilité pour le Gouvernement de rendre obligatoire la dispensation à l'unité de médicaments en rupture.
Enfin, la commission a jugé souhaitable que les leviers d'épargne puissent être utilisés par le Gouvernement en amont de l'apparition de ruptures d'approvisionnement, afin de prévenir leur apparition.
Elle souligne que la commission d'enquête sénatoriale précitée a regretté « une anticipation insuffisante » des ruptures et recommandé d'agir le plus en amont possible lorsque l'existence d'un risque est connue1022(*).
À l'initiative de sa rapporteure, la commission a, en conséquence, adopté un amendement n° 293 permettant au ministre de la santé d'avoir recours à ces mesures lorsqu'un risque de rupture a été déclaré par l'exploitant ou détecté par l'ANSM.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article
34
Faciliter l'inscription à la nomenclature d'un acte associé
à l'utilisation d'un dispositif médical à usage
collectif
Cet article propose de permettre aux exploitants de dispositifs médicaux à usage collectif de demander à la Haute Autorité de santé (HAS) de s'autosaisir sur l'inscription d'un acte ou d'une prestation à la nomenclature. Il vise également à accélérer les décisions de prise en charge en supprimant des délais supplémentaires accordés à la HAS ou au Haut Conseil des nomenclatures au cours de ces procédures.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - Le dispositif proposé
A. Le droit en vigueur : une inscription d'un acte associé à un dispositif médical décorrélée du remboursement du dispositif médical
Pour qu'un dispositif médical puisse être pris en charge par l'assurance maladie, il doit être inscrit à la liste des produits et prestations remboursables (LPPR) prévues à l'article L. 165-1 du code de la sécurité sociale. Cette inscription ne vaut pas prise en charge des actes et prestations associés à ce dispositif médical, qui doivent bénéficier d'une inscription à la classification commune des actes médicaux (CCAM), à la nomenclature générale des actes professionnels (NGAP)1023(*) ou à la nomenclature des actes de biologie médicale (NABM) selon la procédure de droit commun prévue à l'article L. 162-1-7 du code de la sécurité sociale (voir encadré ci-après).
Toutefois, dans l'attente d'un remboursement pérenne, le dispositif médical peut également être inscrit à la prise en charge transitoire (PECT) prévu à l'article L. 165-1-5 du code de la sécurité sociale. Depuis la LFSS pour 20231024(*), lorsque l'utilisation d'un dispositif médical inscrit à la PECT nécessite un acte, ce dernier peut faire l'objet, par dérogation, d'une inscription transitoire à la nomenclature (CCAM, NABM, NGAP) par arrêté, après avis de la Haute Autorité de santé (HAS), des ministres chargés de la santé et de la sécurité sociale. Lorsque le dispositif médical est reconnu sur la LPPR, cette prise en charge transitoire de l'acte est prolongée le temps de son inscription à la nomenclature dans les conditions de droit commun prévues à l'article L. 162-1-7 du code de la sécurité sociale.
Procédure d'inscription d'un acte aux nomenclatures (article L. 162-1-7 du CSS)
1. La demande d'inscription est adressée par l'Union nationale des caisses d'assurance maladie (Uncam) ou par les ministres chargés de la santé et de la sécurité sociale pour avis à la HAS. La demande peut aussi émaner de conseils nationaux professionnels ou d'associations d'usagers agréées.
2. L'évaluation médicale de l'acte est effectuée par la HAS. Elle porte sur l'évaluation du service attendu ou du service rendu de l'acte ou de la prestation qui lui est soumis ainsi que, le cas échéant, sur les actes existants dont l'évaluation pourrait être modifiée en conséquence. À la demande du collège, l'avis de la HAS peut être préparé par une commission spécialisée au sein de la HAS : la commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS). Cet avis est transmis à l'Uncam dans un délai de six mois suivant le dépôt de la demande, renouvelable une fois pour les évaluations complexes.
3. L'évaluation scientifique et technique est de la compétence du Haut Conseil des nomenclatures (HCN) qui est chargé de proposer une méthodologie de description et de hiérarchisation des actes et prestations, ainsi que d'étudier à cette fin ceux qui lui sont soumis. Le HCN établit son rapport en tenant compte des enjeux de pertinence médicale. Ce rapport est ensuite remis, dans un délai de six mois, renouvelable une fois pour les évaluations complexes, à l'Uncam, après avis simple de la commission professionnelle compétente pour la profession de médecin. Il remet chaque année un rapport annuel d'activité après consultation de l'ensemble des acteurs impliqués dans la hiérarchisation.
En outre, le HCN peut inscrire un acte innovant à la nomenclature pour une période provisoire de trois ans, renouvelable une fois.
4. La tarification relève de la compétence de l'Uncam en fonction de la hiérarchisation.
5. Consultation. L'Uncam sollicite l'avis de l'Union nationale des organismes complémentaires d'assurance maladie (Unocam) et, le cas échéant, de la HAS lorsque la décision porte sur l'évaluation du service attendu ou du service rendu d'un acte ou d'une prestation.
6. Inscription. Les conditions d'inscription d'un acte ou d'une prestation, leur inscription et leur radiation sont décidées par l'Uncam. Les décisions d'inscription de l'Uncam sont réputées approuvées sauf opposition motivée des ministres chargés de la santé et de la sécurité sociale dans un délai de 21 jours.
Source : Rapport n° 99, tome II (2022-2023) de la commission des affaires sociales du Sénat, p. 283
B. Le dispositif proposé
1. La procédure d'inscription des actes associés à un dispositif médical
La procédure de demande d'inscription d'un acte aux nomenclatures peut être engagée à l'initiative du ministre chargé de la santé, de l'Uncam ou bien du conseil national professionnel (CNP) concerné par l'acte ou d'une association d'usagers. Lorsque les demandes de saisine émanent d'un CNP ou d'usagers, le dossier n'est pas directement intégré au programme de travail de la Haute Autorité mais fait l'objet d'une sélection (voir encadré ci-après). La HAS précise que le nombre de demandes via des associations d'usagers sont rares : une demande par an tout au plus. Plus prolifique, le canal des CNP reste néanmoins variable selon les années. Au cours des six ans passés, la HAS relève entre huit et vingt-deux demandes déposées par an découlant sur trois à huit demandes acceptées par an - soit un taux d'acceptation variant de 31 à 57 %.
Procédure de sélection des demandes d'inscription par la HAS
Pour les demandes émanant des CNP ou des associations d'usagers, une procédure spécifique a été mise en place en 2017 et actualisée en 2022. Le service de l'évaluation des actes professionnels (SEAP) de la HAS reçoit la demande via la plateforme EvActe, identifie l'objectif de l'évaluation à mener et analyse les besoins à couvrir par l'acte à évaluer et la faisabilité de l'évaluation à mener. Un groupe de priorisation s'attelle par la suite à proposer au collège de la HAS les demandes prioritaires. Le collège décide ensuite des dossiers à inscrire au programme de travail de la Haute Autorité.
Les demandes portant sur un acte associé à un dispositif médical inscriptible à la LPPR font l'objet d'une autre procédure et sont évalués conjointement avec le dispositif médical. Saisie d'une demande d'inscription d'un dispositif médical, la CNEDiMTS peut s'autosaisir sur l'acte associé lorsqu'aucun acte ne correspond à l'utilisation du dispositif médical.
En revanche, pour les actes non associés à des dispositifs médicaux inscriptibles à la LPPR, la HAS souligne que « par définition, nous n'avons pour le moment que peu de dossiers qui ont trait à des industriels ». Depuis 2017, 115 évaluations de demandes ont effectivement été réalisées dont seulement 7 (soit 6 %) sont des dossiers déposés par des industriels ou déposés par d'autres acteurs en relais d'une demande industrielle. Il s'agit pour l'instant d'évaluations à la suite d'un forfait innovation, d'évaluations de tests compagnons, d'actes diagnostiques avec dépôt par le CNP ou institution en relais d'une demande industrielle. Il n'y a pas encore eu d'évaluation d'acte thérapeutique ayant trait à une demande d'industriel.
Source : Procédure et réponses de la HAS au questionnaire de la rapporteure
• Le présent article - au 3° - modifie l'article L. 162-1-7 précité afin de permettre aux exploitants de dispositifs médicaux ou de dispositifs médicaux de diagnostic in vitro de demander à la HAS de s'autosaisir sur l'inscription d'un acte ou d'une prestation aux nomenclatures lorsque le dispositif médical est à usage collectif et qu'il est associé soit à l'action thérapeutique soit à l'action diagnostique de l'acte à évaluer.
Selon l'étude d'impact, il s'agit ainsi de permettre aux industriels « d'accéder directement au formulaire de demande d'évaluation d'acte professionnel actuellement utilisé par les CNP, via la plateforme EVActe de la HAS ». Cette demande des industriels devra toutefois « spécifiquement contenir un courrier de soutien du CNP directement concerné par l'acte à évaluer »1025(*). Le CEPS note également que « le système actuel avec l'impossibilité pour les exploitants de faire une demande de création d'acte alors que leur produit en nécessite un ren[d] plus longue et plus opaque l'inscription des nouveaux produits ».
2. La suppression des délais supplémentaires lors du processus d'évaluation
L'article L. 162-1-7 du code de la sécurité sociale permet une prise en charge provisoire des actes innovants, c'est-à-dire des actes ayant bénéficié d'une amélioration de service attendu (ASA) de niveau I à III, soit une amélioration considérée comme majeure, importante ou modérée.
• Le 1° du présent article propose de supprimer le critère tenant au caractère innovant des actes pour leur inscription sur la liste provisoire par le HCN. Il étend donc la prise en charge provisoire aux actes ayant bénéficié d'une ASA IV (amélioration mineure) et V (absence d'amélioration). En outre, il supprime la période renouvelée de trois ans pouvant être accordée à une inscription sur la liste provisoire.
• Le 2° vise à supprimer le délai supplémentaire de six mois accordé, lorsque les évaluations sont complexes :
- au collège de la HAS pour la remise à l'Uncam de l'avis relatif à l'inscription de l'acte ;
- au HCN pour la remise à l'Uncam du rapport relatif à la description et à la hiérarchisation de l'acte ou de la prestation concerné.
Cette suppression vise à réduire le délai s'écoulant de la saisine de la HAS jusqu'à l'entrée en vigueur de la décision d'inscription, lequel, selon l'étude d'impact, peut atteindre un an et demi1026(*).
II - Les modifications considérées comme adoptées par l'Assemblée nationale
Inséré au texte, un amendement de la rapporteure générale Stéphanie Rist revient sur la suppression du délai renouvelable de trois ans pour l'inscription à la liste provisoire d'un acte. En lieu et place, il est proposé de maintenir une possibilité de renouvellement de 18 mois. Selon l'objet de l'amendement, « cette possibilité est en effet parfois nécessaire pour disposer de données complémentaires ».
Un second amendement retenu de la rapporteure générale propose une clarification d'ordre rédactionnel.
Cet article est considéré comme ayant été adopté par l'Assemblée nationale, ainsi modifié.
III - La position de la commission
La rapporteure accueille favorablement les dispositions du présent article proposant d'introduire une procédure de demande d'inscription d'un acte à la main des exploitants de dispositifs médicaux. Cette procédure permettra une meilleure lisibilité de la procédure.
En outre, la rapporteure partage l'intention du Gouvernement d'accélérer les délais d'inscription d'un acte aux nomenclatures. La HAS n'est cependant pas favorable à la suppression du délai supplémentaire accordé lorsque l'évaluation de l'acte est complexe. Le formulaire de demande d'inscription de l'acte, déposé auprès de la HAS, s'avère souvent incomplet. Dès lors, la Haute Autorité note que « l'évaluation des actes requiert ainsi systématiquement une étape de cadrage de la demande [...], dans un format correspondant aux standards internationaux et européens, puis une analyse critique exhaustive de la littérature pouvant nécessiter la réalisation de méta-analyse, ou d'analyse spécifique de données, la constitution d'un groupe d'experts (professionnels de santé et patients) pour l'ensemble des spécialités concernées et sans conflit d'intérêt, la consultation systématique des parties prenantes (CNP, association de patients) correspondant à une phase contradictoire intégrée dans la procédure, avant l'étape de validation ». Toujours selon la HAS, cette méthode générale, conforme aux exigences les plus élevées, ne peut être respectée en tenant un délai de six mois.
La rapporteure partage le sentiment selon lequel la suppression du délai supplémentaire pourrait s'avérer contre-productif en incitant la Haute Autorité à rejeter davantage de dossiers faute de temps pour réunir les données nécessaire à l'expertise. C'est pourquoi, la commission a adopté un amendement n° 295 de sa rapporteure afin de maintenir ce délai complémentaire de six mois.
S'agissant de l'inscription à la liste provisoire des actes professionnels, la rapporteure souscrit à l'extension de cette prise en charge temporaire aux actes bénéficiant d'une ASA moindre. Comme le note la HAS, « pour ces derniers, le remboursement provisoire permettrait d'encourager le recueil de données complémentaires. Ces données pourraient ensuite alimenter une réévaluation par la HAS à l'issue de la période de remboursement provisoire et ainsi éclairer la Cnam sur la pertinence de rembourser de manière pérenne ou de ne plus rembourser l'acte considéré ».
En revanche, la rapporteure n'est pas favorable à la suppression, prévue dans le texte initial, du renouvellement du délai de trois ans pour l'inscription provisoire d'un acte. Si la rapporteure prend acte de l'amendement de Stéphanie Rist, retenu dans le texte transmis au Sénat, conservant une possibilité de renouveler la période d'inscription provisoire pour un délai de 18 mois, elle estime toutefois que la période totale qui en résulterait - quatre ans et demi - demeurerait insuffisante pour le recueil des données nécessaires à la réévaluation de l'acte à inscrire. Interrogée sur ce point, la HAS a mentionné l'importance de disposer d'un délai de six ans pour collecter les données pertinentes, notamment en vie réelle. La rapporteure a donc proposé un amendement n° 294, adopté par la commission, permettant de maintenir le renouvellement possible pour une période de trois ans de la prise en charge provisoire.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article
34 bis (nouveau)
Recueil des données d'efficacité visant
à évaluer la performance des médicaments de
thérapie innovante (MTI)
Cet article, inséré par le Gouvernement dans le texte sur lequel il a engagé sa responsabilité en application du troisième alinéa de l'article 49 de la Constitution, vise à clarifier les modalités de recueil des données en vie réelle nécessaires au financement des médicaments de thérapie innovante et à permettre la mobilisation de bases de données publiques.
La commission propose d'adopter cet article sans modification.
I - Le dispositif proposé
A. Le modèle de financement des médicaments de thérapie innovante mis en place par la LFSS pour 2023
Quatre types de médicaments de thérapie innovante (MTI) sont distingués en droit européen : les médicaments de thérapie génique, les médicaments de thérapie cellulaire somatique, les produits issus de l'ingénierie tissulaire et les MTI combinés, appartenant à l'un des trois types précédents mais incorporant un ou plusieurs dispositifs médicaux1027(*).
Onéreux, les MTI sont, pour l'essentiel, dispensés à l'hôpital et pris en charge en dehors des groupes homogènes de séjour (GHS) de la tarification à l'activité, par leur inscription sur la liste en sus1028(*).
Afin d'alléger les contraintes pesant sur la trésorerie des hôpitaux et pour maîtriser le risque financier attaché au financement de ces médicaments, la LFSS pour 20231029(*) a mis en place des modalités de financement dérogatoires des MTI permettant de faire porter une partie des flux financiers directement par l'assurance maladie et de subordonner le paiement aux résultats obtenus en vie réelle.
Désormais, le code de la sécurité sociale1030(*) prévoit que, lorsque le prix demandé par un exploitant pour un MTI inscrit sur la liste en sus est supérieur à un seuil défini par arrêté, le coût du traitement est déterminé par convention ou, à défaut, par décision du Comité économique des produits de santé (CEPS).
Un arrêté fixe, par ailleurs, un « forfait de thérapie innovante » correspondant au montant maximal que les hôpitaux peuvent décaisser pour l'acquisition d'un MTI. Lorsque le coût défini conventionnellement ou par décision du CEPS est supérieur à ce forfait, son règlement est réalisé directement par l'assurance maladie, en un ou plusieurs virements annuels.
Les modalités de ce paiement échelonné, soit le nombre, les montants, les conditions et les échéances des versements réalisés par l'assurance maladie, sont fixées par la convention conclue avec le CEPS ou par la décision de celui-ci. Les paiements doivent tenir compte des données d'efficacité en vie réelle du médicament concerné et sont interrompus en cas d'échec du traitement.
Le code de la sécurité sociale1031(*) prévoit que l'exploitant assure à sa charge le recueil des données permettant d'évaluer l'efficacité en vie réelle du traitement et qu'à cette fin, les prescripteurs lui transmettent les données de suivi des patients traités.
B. L'article 34 bis vise à préciser les modalités de recueil des données en vie réelle
L'article 34 bis, issu d'un amendement du Gouvernement retenu dans le texte sur lequel ce dernier a engagé sa responsabilité, vise à clarifier les modalités de recueil des données en vie réelle fondant le nouveau système de financement des MTI et à permettre, notamment, l'utilisation de bases de données publiques par les exploitants.
Pour ce faire, il modifie l'article L. 162-16-6 du code de la sécurité sociale pour prévoir :
- l'intervention d'un décret précisant les modalités de recueil, par les prescripteurs, des données de suivi des patients traités ;
- la transmission de ces données, dans des conditions assurant le respect du secret médical, au CEPS et à l'entreprise assurant l'exploitation, l'importation ou la distribution parallèle du médicament ;
- que l'entreprise participe en tout ou partie au financement du recueil des données.
D'après le Gouvernement, les dispositions actuelles peuvent « empêcher la mobilisation de bases de données publiques existantes, et induire une difficulté de transmission des données d'intérêt aux administrations concernées, dont le CEPS et l'assurance maladie »1032(*).
Au contraire, les présentes dispositions devraient permettre la fixation par décret des modalités de recueil des données, qui pourront autoriser ou imposer l'utilisation d'outils existants, et prévoient explicitement la transmission de ces données au CEPS.
II - La position de la commission
La commission avait soutenu, lors de l'examen du PLFSS pour 2023, la mise en place de ce nouveau dispositif de financement des MTI, jugeant que celui-ci devrait permettre de maîtriser le risque financier attaché à ces innovations1033(*).
Elle demeure convaincue, un an plus tard, qu'un financement fondé sur l'efficacité mesurée des traitements en vie réelle est d'autant plus indispensable que les innovations thérapeutiques apparaissent de plus en plus coûteuses. La croissance rapide des dépenses brutes associées à la liste en sus, avant application des remises conventionnelles comme de la clause de sauvegarde, en fournit une illustration éclairante.
Évolution des dépenses brutes de produits de santé de la liste en sus
(en millions d'euros)

Source : Commission des affaires sociales du Sénat, d'après des données CCSS
En conséquence, la commission a favorablement accueilli les dispositions du présent article, qui devraient permettre de faciliter l'utilisation de bases de données existantes dans le recueil des données nécessaires à l'évaluation des médicaments. Elle a également jugé nécessaire que ces données soient accessibles au CEPS comme à la Caisse nationale de l'assurance maladie (Cnam) et aux administrations concernées.
La commission propose d'adopter cet article sans modification.
Article
35
Améliorer les dispositifs d'accès dérogatoires aux
produits de santé
Cet article propose diverses adaptations aux dispositifs d'accès précoce et d'accès compassionnel.
Il vise à créer un régime dérogatoire de prise en charge pour des médicaments dont la prise en charge au titre de l'accès précoce a pris fin à la suite de l'attribution, par la HAS, d'une amélioration de service médical rendu V (ASMR V) ou d'un service médical rendu (SMR) suffisant en l'attente de données supplémentaire. Il vise également à adapter les modalités de l'accès précoce aux vaccins, à garantir un approvisionnement adapté pour les produits bénéficiant de l'accès précoce et à mieux articuler les dispositifs d'accès précoce et d'accès compassionnel.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - Le dispositif proposé
A. Les modalités de prise en charge de droit commun pour les médicaments
1. L'autorisation de mise sur le marché, préalable à la commercialisation de tout médicament
Avant de pouvoir être administré aux patients dans une indication, y compris à titre gratuit, un médicament fait l'objet d'une évaluation de son rapport bénéfices/risques qui donne lieu, si elle est concluante, à une autorisation de mise sur le marché (AMM). Sont notamment pris en compte, pour le décernement d'une AMM, le degré d'efficacité du produit, ses effets indésirables prévisibles, la qualité chimique du produit et celle des procédés de fabrication1034(*). La demande d'AMM est à l'initiative de l'industriel exploitant.
Pour accéder au marché français, l'AMM peut être attribuée au niveau européen selon trois procédures, par l'agence européenne du médicament (EMA), en application d'un règlement européen1035(*), ou, à défaut, par l'agence nationale de sécurité du médicament et des produits de santé (ANSM), en application de l'article L. 5121-8 du code de la santé publique. L'ANSM peut également accorder une AMM à un médicament autorisé dans un autre État partie à l'accord sur l'espace économique européen (EEE) en l'absence de demande de l'industriel, si des raisons de santé publique le justifient1036(*).
L'autorisation de mise sur le marché est valable pour une durée initiale de cinq ans, puis peut être renouvelée pour cinq ans ou sans limitation de durée1037(*). Elle devient toutefois caduque au bout de trois ans en l'absence de commercialisation effective, et peut être retirée à tout moment si le médicament est nocif, ne permet pas d'obtenir des résultats thérapeutiques, si le titulaire ne respecte pas ses obligations, si la spécialité n'a pas la composition déclarée et si le rapport entre les bénéfices et les risques devient défavorable1038(*).
Le cas échéant, l'ANSM peut exiger du titulaire de l'AMM des études complémentaires concernant la sécurité, le risque ou l'efficacité du produit autorisé1039(*). Le titulaire est également tenu d'informer l'ANSM et l'EMA sans délai en cas d'engagement d'une action tendant à suspendre ou arrêter la commercialisation d'un médicament bénéficiant d'une AMM ou à solliciter le retrait ou le non-renouvellement d'une AMM1040(*).
2. Pour être pris en charge par la sécurité sociale, le produit doit également faire l'objet d'une procédure complémentaire
L'octroi d'une AMM pour une indication n'implique pas immédiatement l'admission au remboursement, qui fait l'objet d'une procédure spécifique.
Après avoir reçu une AMM, l'exploitant d'un médicament doit en effet saisir la commission de transparence de la Haute Autorité de santé (HAS) afin qu'elle procède à l'évaluation scientifique et médico-économique1041(*) des produits concernés, selon deux principaux axes : l'amélioration du service médical rendu (ASMR) et le service médical rendu (SMR).
Le service médical rendu est un indicateur composite prenant notamment en compte l'efficacité d'un produit, ses effets indésirables et la gravité de la pathologie pour laquelle il est indiqué afin de déterminer si une prise en charge par la solidarité nationale est justifiée. Le niveau de SMR détermine l'admissibilité au remboursement et, le cas échéant, le taux de remboursement par la sécurité sociale pour un médicament remboursable. La HAS, par le biais de sa commission de transparence, fixe le niveau de SMR d'un produit de santé, qui peut être majeur ou important, modéré ou faible mais justifiant cependant le remboursement, ou insuffisant pour justifier une prise en charge par la collectivité1042(*).
L'amélioration du service médical rendu vise à évaluer le progrès thérapeutique apporté par un médicament, en comparaison aux traitements déjà disponibles. Il existe cinq niveaux d'ASMR, allant d'ASMR I (majeure) à ASMR V (inexistante). Le niveau d'ASMR, fixé par la commission de transparence de la HAS, influe sur la détermination du prix du médicament.
La procédure varie ensuite entre les médicaments dispensés en officine et à l'hôpital.
a) La fixation du prix et des modalités de prise en charge en ville
En ville, une fois le SMR et l'ASMR du produit
déterminés, et
- lorsque les conséquences
budgétaires attendues et le niveau d'innovation la justifient -
l'évaluation médico-économique d'efficience rendue par la
commission d'évaluation économique et de santé publique de
la HAS réalisée, deux processus prennent place afin de
fixer le prix et le taux de remboursement applicables.
Sur la base de la transmission par la HAS du niveau d'ASMR et, le cas échéant, de l'avis d'efficience, mais aussi du prix des médicaments à même visée thérapeutique, des prix pratiqués à l'étranger et des volumes de vente attendus, le prix du médicament est fixé par négociation entre le comité économique des produits de santé (CEPS) et l'industriel, ou, à défaut, par décision du CEPS.
Sur la base du SMR évalué par la HAS, et si celui-ci est suffisant, l'union nationale des caisses d'assurance-maladie (Uncam) définit le taux de remboursement par la sécurité sociale : 65 % en cas de SMR important, 30 % en cas de SMR modéré et 15 % en cas de SMR faible.
Le ministre de la santé arrête l'inscription du médicament au remboursement, qui fait l'objet d'une publication au Journal officiel.
Circuit simplifié de commercialisation d'un médicament remboursable
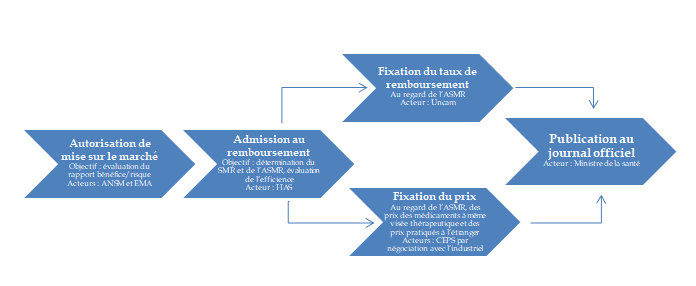
Source : Commission des affaires sociales du Sénat
b) À l'hôpital, trois listes coexistent, déterminant des conditions de prise en charge différenciées
Après détermination de son ASMR et de son SMR, un médicament doit, pour être dispensé à l'hôpital, être inscrit sur la liste des médicaments agréés à l'usage des collectivités mentionnée à l'article L. 5123-2 du code de la santé publique. Un SMR insuffisant fait obstacle à l'inscription d'un médicament sur cette liste.
Pour les médicaments inscrits sur la liste des médicaments agréés à l'usage des collectivités, la fixation du prix est libre, éventuellement dans la limite d'un tarif maximal1043(*). Les médicaments inscrits sur cette liste ne font, en principe, pas l'objet d'une prise en charge spécifique par la sécurité sociale : leur coût est réputé inclus dans le montant qui leur est attribué au titre des groupes homogènes de séjour.
Toutefois, les médicaments inscrits non seulement sur la liste des médicaments agréés à l'usage des collectivités mais aussi sur la liste en sus1044(*) ou la liste de rétrocession1045(*) font l'objet d'une prise en charge de la sécurité sociale et voient leur prix de vente encadré. Celui-ci fait l'objet d'une déclaration au CEPS et d'une publication au Journal officiel.
La liste en sus
Les médicaments inscrits pour une indication sur la liste en sus, mentionnée à l'article L. 162-22-7 du code de la sécurité sociale, bénéficient d'une prise en charge intégrale de la sécurité sociale en sus de la tarification à l'activité (T2A). Leur régime de prise en charge est donc favorable par rapport aux médicaments inscrits sur la seule liste des médicaments agréés à l'usage des collectivités, dont le coût est réputé couvert par la T2A.
Pour être inscrit sur la liste en sus, une indication d'une spécialité pharmaceutique doit être administrée principalement à l'hôpital, présenter un SMR majeur ou important et un ASMR I, II, III ou IV. De plus, son coût doit excéder 30 % du montant du forfait attribué pour le groupement homogène de séjour considéré, et donc représenter un coût excessif pour l'hôpital hors prise en charge en sus des tarifs des GHS.
Ces médicaments sont généralement des produits innovants mais onéreux, que les établissements de santé ne pourraient que difficilement se procurer sans prise en charge financière complémentaire. De nombreux médicaments inscrits sur la liste en sus sont des traitements contre le cancer tels que les immunothérapies.
Les dépenses au titre des médicaments sur la liste en sus sont très dynamiques, avec un taux de croissance annuel moyen de 13,7 % entre 2016 et 2021 : elles dépassent désormais 6 milliards d'euros par an.
B. L'accès précoce et l'accès compassionnel sont deux modalités dérogatoires de prise en charge de médicaments introduits en 2021 en remplacement de dispositifs préexistants
Compte tenu des délais engendrés par la procédure de droit commun - 497 jours s'écoulent, en moyenne, entre l'obtention de l'AMM et l'accès au marché1046(*) - et de l'intérêt thérapeutique de certains produits, des procédures de prise en charge dérogatoires, encadrées par la loi, sont prévues de longue date.
Les dérogations historiques en la matière, l'autorisation temporaire d'utilisation (ATU), créée en 1992, et la recommandation temporaire d'utilisation (RTU), créée en 2014, ne donnaient plus satisfaction : du fait des modifications incrémentales apportées aux dispositifs, ces derniers étaient devenus à la fois complexes, peu lisibles et, pour certains, redondants.
En conséquence, le législateur a remplacé, en loi de financement de la sécurité sociale pour 20211047(*), les six dispositifs d'accès préexistants par deux dispositifs nouveaux permettant une fourniture et une prise en charge dérogatoire de médicaments en établissements de santé, dans un double objectif de lisibilité pour les professionnels et de rapidité de prise en charge pour les patients. Les deux dispositifs nouvellement créés, l'accès précoce et l'accès compassionnel, sont entrés en vigueur le 1er juillet 2021.
Articulation entre les dispositifs dérogatoires historiques et les nouveaux dispositifs d'accès précoce et compassionnel

Source : Ministère de la santé et de la prévention
1. L'accès précoce permet aux patients en impasse thérapeutique et atteints d'une maladie grave une prise en charge intégrale et anticipée pour des produits innovants ayant vocation à être commercialisés
a) L'accès précoce permet aux patients en impasse thérapeutique et atteints d'une maladie grave de bénéficier par anticipation de certains produits encore non autorisés pour leur indication thérapeutique mais ayant vocation à l'être
L'accès précoce, défini à l'article L. 5121-12 du code de la santé publique, est venu en remplacement de l'ATU pour offrir aux patients atteints d'une maladie grave et en impasse thérapeutique un accès anticipé à des médicaments innovants, susceptibles d'être indiqués pour leur pathologie mais non encore autorisés à la mise sur le marché ou pris en charge. Il est principalement placé sous la responsabilité de la HAS1048(*).
L'accès précoce trouve à s'appliquer sous diverses conditions, énumérées à l'article L. 5121-12 précité du code de la santé publique :
- des critères d'accès cumulatifs liés à l'amélioration attendue du médicament sur la santé des patients :
• il n'existe pas de traitement approprié à la pathologie ;
• la mise en oeuvre du traitement ne peut être différée ;
• il existe une forte présomption d'efficacité et de sécurité du médicament ;
• le médicament est présumé innovant ;
• le médicament est indiqué dans une maladie grave, rare ou invalidante ;
- des conditions alternatives liées à la situation du médicament au regard du parcours de prise en charge de droit commun :
• le médicament ne dispose pas d'une AMM pour l'indication concernée, mais l'industriel a déposé ou s'engage à déposer une demande d'AMM dans un délai fixé par la HAS et plafonné à 2 ans1049(*) ;
• le médicament dispose d'une AMM pour l'indication concernée mais n'est pas inscrit sur les listes de remboursement, mais l'industriel a déposé ou s'engage à déposer une demande d'inscription dans le mois suivant l'AMM.
La HAS rend, dans les trois mois1050(*) suivant le dépôt du dossier par l'industriel, sa décision sur l'autorisation de l'accès précoce, valable pour un an éventuellement renouvelable1051(*). Lorsque le médicament ne bénéficie pas d'une AMM pour l'indication concernée, la décision est prise par la HAS après avis conforme de l'ANSM concernant la présomption d'efficacité et de sécurité du médicament1052(*).
L'exploitant d'un produit bénéficiant d'un accès précoce pour une indication s'engage à assurer la continuité des traitements initiés pendant la durée et dans l'année suivant la période de prise en charge précoce.
L'autorisation d'accès précoce prend fin :
- automatiquement dès lors que le médicament est inscrit, pour l'indication considérée, sur la liste des médicaments agréés à l'usage des collectivités, la liste des médicaments remboursables en ville ou la liste de rétrocession ;
- sur arrêté des ministres chargés de la santé et de la sécurité sociale lorsque les conditions d'admission ne sont plus remplies, après suspension ou retrait de l'autorisation décidé par la HAS, le cas échéant sur demande de l'ANSM, notamment en cas d'urgence, si la condition concernée est la présomption forte d'efficacité et de sécurité du médicament ou en cas de non-octroi de l'AMM.
À l'issue de la période d'accès précoce, les spécialités ayant démontré leur efficacité pour une ou plusieurs indications données ont vocation à être prises en charge au titre de la liste en sus.
b) Les médicaments bénéficiant d'une autorisation d'accès précoce sont pris en charge intégralement par la sécurité sociale selon des modalités dérogatoires
Le médicament bénéficiant d'une autorisation d'accès précoce fait l'objet d'une prise en charge intégrale, immédiate et automatique par la sécurité sociale, à titre dérogatoire1053(*), en sus des tarifs hospitaliers1054(*). Le long de la période d'accès précoce, l'indemnité versée par la sécurité sociale en contrepartie de la fourniture du médicament est fixée unilatéralement par l'industriel en cas de primo-indication, ou est fixée au niveau du prix prédéterminé ou du prix maximal de vente aux établissements de santé en cas d'extension d'indication.
Il s'applique toutefois, sur le chiffre d'affaires facturé aux établissements de santé pour chaque indication, un double système de remises régi par l'article L. 152-16-5-1-1 du code de la sécurité sociale :
- une remise annuelle, dont le taux est croissant avec le chiffre d'affaires facturé aux établissements de santé pour le médicament dans l'indication considérée, communiqué tous les 15 février par les industriels. Cette remise peut être majorée, notamment en cas de remise en cause de la présomption d'innovation, d'inscription au remboursement d'une alternative thérapeutique, ou de manquement aux obligations de dépôt de demandes d'AMM ou d'inscription au remboursement dans les délais impartis ;
- une remise de débouclage ayant lieu en fin d'accès précoce et visant à faire correspondre rétroactivement le chiffre d'affaires réalisé par l'industriel sur l'indication considérée avec l'indemnité d'accès précoce, minoré par les remises annuelles, avec celui qui aurait prévalu si la sécurité sociale avait payé, lors de la période d'accès précoce, le prix inscrit au journal officiel1055(*). Ce versement peut avoir lieu sur deux années successives ou en une seule fois, auquel cas une décote peut s'appliquer dans la limite de 3 %.
Entre 2021 et 2023, 125 décisions, dont 98 favorables, ont été rendues par la HAS sur des premières demandes d'accès précoces, bénéficiant au total à plus de 100 000 patients en impasse thérapeutique. Ces patients, pour moitié en oncologie et cancérologie, ont pu bénéficier d'une prise en charge en moyenne 293 jours avant l'inscription des médicaments dans les indications concernées en droit commun, soit plus de neuf mois.
2. L'accès compassionnel vise à autoriser l'utilisation de médicaments ne bénéficiant pas d'une AMM pour une indication considérée et n'ayant pas vocation à être commercialisés pour cette indication
a) Les conditions d'éligibilité à l'accès compassionnel
L'accès compassionnel vise à permettre, exceptionnellement, d'utiliser un médicament dans une indication thérapeutique pour laquelle il ne dispose pas d'une AMM et n'a pas vocation à en recevoir une. Il cible donc des besoins médicaux auxquels peuvent répondre des médicaments pour lesquels un laboratoire n'a pas de stratégie commerciale. L'accès compassionnel est principalement géré par l'ANSM.
Pour bénéficier d'un accès compassionnel, un médicament doit1056(*) :
- répondre à une pathologie pour laquelle il n'existe pas de traitement approprié ;
- ne pas faire l'objet d'une recherche impliquant la personne humaine à des fins commerciales ;
- bénéficier d'une efficacité et d'une sécurité présumées au regard des données cliniques disponibles.
On distingue, au sein de l'accès compassionnel, l'autorisation d'accès compassionnel, qui a succédé aux ATU nominatives, du cadre de prescription compassionnelle, qui a pris la suite des RTU.
b) L'autorisation d'accès compassionnel permet, sur demande d'un prescripteur, de faire bénéficier un patient en impasse thérapeutique nominativement désigné d'un médicament ne disposant d'aucune AMM
L'autorisation d'accès compassionnel (AAC) permet à un patient en impasse thérapeutique de bénéficier, en établissement de santé, d'un médicament ne disposant d'aucune AMM, quelle que soit l'indication thérapeutique. L'autorisation d'accès compassionnel est accordée pour une durée d'un an renouvelable, et, contrairement à l'autorisation d'accès précoce, sur demande d'un prescripteur - et non d'un laboratoire - et pour un patient nominativement désigné - et non pour une indication.
Par dérogation, l'ANSM peut accorder une autorisation d'accès compassionnel dite « pré-précoce » d'un médicament dans une indication qui fait l'objet d'une recherche clinique à un stade très précoce et pour laquelle l'exploitant souhaite obtenir une AMM, si l'entreprise s'engage à déposer une demande d'accès précoce. Cette autorisation d'accès vise à couvrir la période préalable à l'octroi d'une autorisation d'accès précoce : c'est pourquoi certaines conditions pour bénéficier d'un accès précoce s'appliquent pour l'octroi par l'ANSM d'une autorisation d'accès compassionnel pré-précoce. Le patient doit alors être atteint d'une maladie rare, grave ou invalidante pour laquelle le traitement ne peut être différé.
Le refus d'octroi d'un accès précoce à une spécialité pour une indication donnée fait, en principe, obstacle à l'octroi par l'ANSM d'une autorisation d'accès compassionnelle.
c) Le cadre de prescription compassionnelle vise à sécuriser une pratique de prescription sans AMM sur une indication pour un médicament disposant déjà d'AMM dans d'autres indications
Le cadre de prescription compassionnelle (CPC), quant à lui, vise à sécuriser pour une durée de trois ans renouvelable une pratique de prescription sans AMM sur une indication pour un médicament disposant d'une AMM dans d'autres indications. Contrairement à l'autorisation d'accès compassionnel, le cadre de prescription compassionnelle n'est pas nominatif, mais concerne une indication. En outre, il n'est pas accordé à la demande d'un prescripteur, mais à l'initiative de l'ANSM, des ministres de la santé ou de la sécurité sociale ou à la suite d'un signalement. Enfin, il peut, dans certains cas concerner des médicaments dispensés en ville.
d) Le cadre de prise en charge de l'accès compassionnel
Un médicament bénéficiant d'une autorisation d'accès compassionnel ou d'un cadre de prescription compassionnelle fait l'objet d'une prise en charge dérogatoire par la sécurité sociale, aux termes de l'article L. 162-16-5-2 du code de la sécurité sociale. Cette prise en charge est immédiate et automatique, que ce soit dans le cadre d'une AAC ou d'un CPC. Elle se fait, le cas échéant, en sus des tarifs hospitaliers1057(*).
Si la spécialité bénéficiant d'un accès compassionnel fait déjà l'objet d'une prise en charge en ville sur une autre indication, les conditions de prise en charge en accès compassionnel sont identiques à celles qui prévalent pour l'indication concernée. Il en va de même si la spécialité est inscrite sur les listes en sus et de rétrocession.
S'il existe un prix maximal de vente aux établissements de santé fixé pour les médicaments sur la liste des médicaments agréés à l'usage des collectivités publiques, ce prix s'applique également.
Dans le cas contraire, la prise en charge se fait sur la base du prix facturé aux établissements de santé, déterminé librement par le laboratoire, ou d'une base forfaitaire annuelle par patient.
Comme pour l'accès précoce, des remises annuelles sont versées par les industriels en fonction d'un barème croissant avec le chiffre d'affaires hors taxes réalisé sur chaque indication. Ce système de remises n'est toutefois pas applicable si l'indication est prise en charge sur une base forfaitaire.
B. Le dispositif proposé vise à adapter le cadre de l'accès précoce et de l'accès compassionnel pour répondre à quatre problématiques récurrentes
1. L'établissement d'une prise en charge dérogatoire pour les médicaments en fin d'accès précoce présentant un ASMR V ou un SMR suffisant en l'attente de données supplémentaires
À l'issue de la période d'accès précoce pour une indication, l'attribution par la HAS d'un SMR modéré ou faible ou un ASMR V en l'attente de données supplémentaires fait obstacle à l'inscription de la spécialité concernée sur la liste en sus : les médicaments concernés ne peuvent alors être inscrits que sur la liste des médicaments agréés à l'usage des collectivités pour l'indication considérée.
L'inscription de ces spécialités sur la seule liste des médicaments agréés à l'usage des collectivités constitue, pour les patients traités, un risque de rupture de traitement compte tenu des tarifs des médicaments concernés, souvent trop onéreux pour que les établissements puissent les prendre en charge en l'absence de financement complémentaire à la T2A.
Pourtant, il est fréquent que la HAS modifie, à l'aune des données supplémentaires, le SMR ou l'ASMR accordé : selon la réponse de la Haute autorité au questionnaire de la rapporteure, pour 2022, une telle réévaluation a eu lieu dans 58 % des cas.
Afin d'éviter une perte de chances pour les patients concernés, le 2° du II de l'article 35 du PLFSS pour 2024 se propose de créer un régime temporaire de prise en charge dérogatoire pour les médicaments en fin d'accès précoce et non-inscrits sur la liste en sus du fait de l'attribution par la HAS d'un ASMR V ou d'un SMR modéré ou faible en l'attente de données supplémentaires.
Le régime de ces nouvelles modalités de prise en charge est défini dans un nouvel article L. 162-16-5-1-2 du code de la sécurité sociale.
Le I et le II de cet article L. 162-16-5-1-2 nouveau précisent que les spécialités concernées par le régime de prise en charge temporaire sont celles :
- réservées à un usage hospitalier (1° du II) ;
- dont la prise en charge au titre de l'accès précoce a pris fin suite à leur inscription sur la liste des médicaments agréés à l'usage des collectivités, la liste des médicaments remboursables en officine ou la liste de rétrocession (I) ;
- ne bénéficiant pas d'une inscription sur la liste en sus (2° du II) ;
- ayant fait l'objet d'un avis de la commission de transparence reconnaissant à la spécialité dans l'indication concernée un SMR ou un ASMR au-dessus d'un plafond fixé par décret, et dont un plan de développement de la spécialité existant pourrait fournir des données supplémentaires susceptibles d'engendrer une actualisation des niveaux de SMR et d'ASMR attribués (3° du II).
Les conditions précitées sont cumulatives.
Le 1° du III de l'article L. 162-16-5-1-2 nouveau du code de la sécurité sociale définit les conditions de prise en charge au titre de l'accès temporaire et dérogatoire. Celle-ci est calculée sur la base d'une indemnité correspondant au prix ou tarif le plus bas dans d'autres États européens comparables1058(*) ou, à défaut, de l'indemnité d'accès précoce, auxquelles est appliquée une décote tenant compte de l'avis de la commission de transparence. La décote est fixée par un arrêté des ministres chargés de la santé et de la sécurité sociale déterminant les éléments à prendre en compte pour sa fixation, notamment le délai laissé à l'industriel pour présenter ses données supplémentaires.
Le IV et le VIII de l'article L. 162-16-5-1-2 nouveau du code de la sécurité sociale rendent applicable au régime de la prise en charge temporaire et dérogatoire les systèmes de remise annuelle (A du IV) et de remise de débouclage prévus pour l'accès précoce (A du IV et VIII). Sans qu'ils ne puissent conduire les reversements à excéder une part minimale du chiffre d'affaires réalisé sur la spécialité, les taux de remise peuvent, en outre, faire l'objet de majorations reconductibles (B) :
- à compter de la deuxième année dans le dispositif ;
- en cas de dépassement du délai laissé à l'exploitant pour présenter à la commission de transparence des données supplémentaires ;
- en l'absence de signature d'une convention fixant le tarif de responsabilité, appliqué pour la prise en charge en sus des tarifs hospitaliers, dans les 180 jours suivant la demande d'inscription sur la liste en sus.
Pour la détermination de la base sur laquelle s'appliquent les remises, les exploitants sont tenus de transmettre au CEPS, le 15 février de chaque année, le chiffre d'affaires réalisé sur chaque indication pour lesquelles une spécialité bénéfice de la prise en charge temporaire, et le nombre d'unité fournies, aux termes du 2° du III de l'article L. 162-16-5-1-2 nouveau du code de la sécurité sociale.
Le V de l'article L. 162-16-5-1-2 nouveau du code de la sécurité sociale précise que la prise en charge temporaire est accordée, sur demande de l'entreprise exploitant le médicament et pour chaque indication, par un arrêté conjoint des ministres de la santé et de la sécurité sociale.
Le VI de l'article L. 162-16-5-1-2 nouveau du code de la sécurité sociale appliquent à la prise en charge temporaire les obligations s'imposant aux exploitants bénéficiaires d'une autorisation d'accès précoce concernant la collecte de données sur l'efficacité du traitement, ses effets indésirables, ses conditions réelles d'utilisation et les caractéristiques des patients traités.
Le VII de l'article L. 162-16-5-1-2 nouveau du code de la sécurité sociale précise que la prise en charge temporaire dérogatoire prend fin :
- au-delà d'une durée fixée par décret qui ne peut dépasser trois ans (4°) ;
- à l'inscription de la spécialité sur la liste en sus (1°) ;
- lorsqu'un nouvel avis de la commission de transparence ne permet pas de fixer le SMR et/ou l'ASMR à un niveau assez élevé pour permettre son inscription sur la liste en sus, sur arrêté des ministres chargés de la santé et de la sécurité sociale (2°) ;
- en cas de retrait de l'AMM (3°) ;
- en cas de radiation de la liste des médicaments agréés à l'usage des collectivités (3°) ;
- en cas d'absence ou de retrait de la demande d'inscription à la liste en sus (3°).
Le IX de l'article L. 162-16-5-1-2 nouveau du code de la sécurité sociale renvoie à un décret en Conseil d'État la détermination de ses conditions d'application.
Le c du 1° du II de l'article 35 du PLFSS pour 2024 précise qu'en cas de prise en charge temporaire et dérogatoire, la remise de débouclage prévue en fin d'accès précoce a lieu à la fin de la période de prise en charge temporaire et prend en compte l'ensemble des périodes de prise en charge, qu'elle soit au titre de l'accès compassionnel pré-précoce, de l'accès précoce ou de la prise en charge temporaire et dérogatoire. Il modifie, pour cela, l'article L. 162-16-5-1-1 du code de la sécurité sociale, relatif aux conditions de prise en charge de l'accès précoce, en rétablissant son V, devenu VI en application du b du 1° du II.
Les 3°, 4° et 5° du II de l'article 35 du PLFSS pour 2024 procèdent à des coordinations juridiques permettant aux articles renvoyant à l'accès précoce de renvoyer également à l'accès temporaire dérogatoire.
Le 3° applique à l'accès dérogatoire et temporaire la déclaration obligatoire de l'indication pour laquelle une AAP, une AAC ou un CPC est prescrit, mentionnée à l'article L. 162-16-5-3 du code de la sécurité sociale. Le 4° fixe le principe d'une prise en charge en sus des tarifs hospitaliers au titre de l'accès temporaire, en modifiant l'article L. 162-22-7-3 du code de la sécurité sociale. Le 5° dispose que les conventions conclues au titre des spécialités ayant bénéficié d'un accès temporaire et dérogatoire n'incluent que des remises portant sur les unités vendues à compter de la signature de la convention. Il modifie pour cela l'article L. 162-18 du code de la sécurité sociale.
L'étude d'impact estime à 39 millions d'euros l'impact financier de ces dispositions pour 2024.
2. L'adaptation de l'accès précoce aux cas particuliers des vaccins
Selon l'étude d'impact annexée par le Gouvernement, les critères de l'accès précoce ne sont pas pleinement adaptés aux vaccins, pour lesquels la HAS émet des recommandations vaccinales aujourd'hui non prises en compte dans l'attribution de l'accès précoce.
Le a du 1° du I de l'article 35 du PLFSS modifie l'article L. 5121-12 du CSP afin d'intégrer les recommandations vaccinales émises par la HAS dans les conditions d'éligibilité à l'accès précoce pour les vaccins.
L'étude d'impact estime à 14 millions d'euros par an l'impact financier de cette réforme.
3. La garantie d'un approvisionnement adapté pour les médicaments en accès précoce
Alors que l'autorisation d'accès précoce est octroyée sur la base d'un dossier de demande comprenant notamment des prévisions de volumes de vente, des limitations de production ont pu être constatées sur des médicaments bénéficiant d'une autorisation d'accès précoce.
En ce sens, le b du 1° du I de l'article 35 vise à renforcer les obligations des industriels en matière d'approvisionnement de leurs produits bénéficiant d'un accès précoce, en contrepartie de la prise en charge dérogatoire et anticipée desdits produits.
Il modifie l'article L. 5121-12 du code de la santé publique, définissant le cadre de l'accès précoce, afin de préciser que l'autorisation d'accès précoce est subordonnée au respect d'un engagement d'approvisionnement approprié et continu du marché national, de manière à couvrir les besoins des patients en France.
Le manquement à cette obligation nouvellement créée fait l'objet d'une sanction financière, définie au a du 1° du II de l'article 35. Celui-ci modifie le B de l'article L. 162-16-5-1-1 du code de la sécurité sociale régissant les cas de majoration des taux de remise annuels pour les médicaments bénéficiant de l'accès précoce, en ajoutant un nouveau motif de majoration en cas de manquement à l'engagement d'approvisionnement. Cette majoration de la remise ne peut avoir lieu qu'après que l'exploitant a été mis en mesure de présenter ses observations.
4. La définition d'une meilleure articulation entre accès précoce et accès compassionnel
Le refus d'accorder une autorisation d'accès précoce à un médicament pour une indication fait aujourd'hui obstacle à l'octroi ultérieur d'une autorisation d'accès compassionnel pour cette même indication.
Toutefois, alors que des médicaments doivent être présumés innovants pour pouvoir bénéficier d'une autorisation d'accès précoce, cette condition n'est pas requise pour bénéficier d'une autorisation d'accès compassionnel. Cela conduit l'ANSM à refuser des autorisations d'accès compassionnel du fait d'un refus d'accès précoce pris sur la seule base de l'absence de caractère innovant présumé d'un produit, alors même que cette condition n'est pas nécessaire dans l'absolu.
Selon l'étude d'impact au PLFSS annexée par le Gouvernement, deux spécialités se seraient vues refuser l'accès précoce au seul motif de l'absence de présomption de caractère innovant depuis l'entrée en vigueur du dispositif au 1er juillet 2021.
Afin de mieux articuler l'octroi des autorisations d'accès précoce et compassionnel, le 2° du I de l'article 35 complète l'article L. 5121-12-1, qui définit l'accès compassionnel et ses conditions d'octroi, par un alinéa précisant que le refus d'octroyer une autorisation d'accès précoce dans une indication sur le seul fondement de l'absence de caractère innovant présumé du médicament ne fait pas obstacle à l'octroi, par l'ANSM, d'une autorisation d'accès compassionnel pour la même indication.
L'étude d'impact estime à 17,6 millions d'euros le coût de cette mesure pour 2024, puis 20 millions d'euros pour les années suivantes.
II - Les modifications considérées comme adoptées par l'Assemblée nationale
Le Gouvernement a retenu, dans le texte sur lequel il a engagé sa responsabilité à l'Assemblée nationale, dix-neuf amendements rédactionnels.
Cet article est considéré comme ayant été adopté par l'Assemblée nationale, ainsi modifié.
III - La position de la commission
La commission a souscrit à l'ensemble des dispositions portées par cet article, qui lui semblent contribuer utilement à résoudre certaines faiblesses des régimes juridiques de l'accès précoce et de l'accès compassionnel.
En particulier, la commission accueille favorablement la mise en place d'un régime de prise en charge temporaire pour les spécialités n'ayant pas pu être inscrites sur la liste en sus du fait de l'attribution d'un ASMR V ou d'un SMR moyen ou faible dans l'attente de données supplémentaires. Compte tenu du fort taux de réévaluations de l'ASMR et/ou du SMR par la HAS une fois les données complémentaires disponibles, qui atteint 58 % en 2022, il semble en effet profondément regrettable de mettre en danger la continuité du traitement administré aux patients faute de possibilités de prise en charge.
La commission se montrera néanmoins attentive au niveau des décotes fixées pour la prise en charge temporaire : celles-ci doivent bien entendu être plus basses lorsqu'elles s'appliquent sur un prix dans les pays comparables que lorsqu'elles amputent une indemnité d'accès précoce.
La commission appelle le Gouvernement à retenir une solution équilibrée pour les paramètres de calcul du niveau de l'indemnité de prise en charge temporaire : s'il est certain que cette indemnité doit, en quelque sorte, sanctionner les résultats insuffisants obtenus par le médicament lors de sa première évaluation par la commission de transparence, il n'est pas moins évident qu'une prise en charge insuffisante priverait le dispositif de toute attractivité, ce qui nuirait, en dernier ressort, aux patients.
En ce sens, la commission a adopté un amendement n° 297 retenant une solution plus équilibrée pour la fixation de l'indemnité d'accès temporaire, définie comme une fraction de l'indemnité d'accès précoce qui lui a été appliquée. Cet amendement supprime donc, pour le calcul de l'indemnité d'accès temporaire, la référence au prix européen le plus bas, qui aurait conduit à des tarifs trop peu compétitifs.
La commission a également adopté les deux amendements rédactionnels nos 296 et 298.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article
36
Soutien au maintien sur le marché des médicaments
matures
Cet article propose une obligation de recherche de repreneurs par une entreprise cessant la commercialisation d'un médicament d'intérêt thérapeutique majeur et, à défaut de reprise ou d'alternative, une possible concession de l'exploitation à un établissement pharmaceutique public.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés visant notamment à :
- permettre une meilleure anticipation d'une éventuelle reprise par un établissement pharmaceutique public ;
- accompagner le renforcement du rôle de l'ANSM dans la supervision des plans de gestion des pénuries pour les médicaments à risque de rupture.
I - Le dispositif proposé
A. Une obligation de recherche de repreneur en cas d'arrêt de commercialisation d'un médicament d'intérêt thérapeutique majeur
Dans le cadre des mesures de lutte contre les pénuries de médicaments, le Gouvernement propose à l'article 36 un dispositif visant à prévenir, pour les médicaments d'intérêt thérapeutique majeur (MITM) aux brevets expirés, des arrêts de commercialisation sans prévoir de solution permettant, par la suite, de couvrir les besoins.
Il modifie à cette fin l'article L. 5124-6 du code de la santé publique, qui prévoit les obligations déclaratives des entreprises pharmaceutiques dans le cas d'une suspension ou d'un arrêt de commercialisation d'un médicament. Il y est ainsi notamment prévu une obligation d'information de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) au moins un an avant la date envisagée pour les MITM, au plus tard deux mois avant la suspension ou l'arrêt pour un autre médicament.
Article L. 5111-4 du code de la santé publique
On entend par médicaments ou classes de médicaments d'intérêt thérapeutique majeur les médicaments ou classes de médicaments pour lesquels une interruption de traitement est susceptible de mettre en jeu le pronostic vital des patients à court ou moyen terme, ou représente une perte de chance importante pour les patients au regard de la gravité ou du potentiel évolutif de la maladie.
Le dispositif s'inspire directement de la loi « Florange »1059(*) adoptée en 2014 et visant à contraindre une entreprise de plus de 1 000 salariés souhaitant fermer un site industriel à chercher un repreneur.
Le premier alinéa du nouveau II ajouté à l'article L. 5125-6 du CSP complète l'obligation d'information déjà prévue au même article dans le cas d'une décision de suspension ou d'arrêt de commercialisation d'un MITM ne faisant plus l'objet d'une protection au titre des droits de la propriété intellectuelle ou industrielle. L'entreprise exploitant le médicament serait alors tenue d'informer l'ANSM des incidences prévisibles de cette suspension ou de cet arrêt sur la couverture des besoins de la population française.
Le deuxième alinéa du même II prévoit l'obligation pour le titulaire de l'autorisation de mise sur le marché (AMM) de rechercher un repreneur dans le cas d'une absence d'alternatives thérapeutiques disponibles permettant de couvrir les besoins de manière pérenne1060(*). Cette situation, constatée par l'ANSM, est notifiée au titulaire de l'autorisation.
Le présent article prévoit ainsi une contrainte formulée comme une obligation de moyens et non de résultats.
Trois modalités sont alors prévues par des 1° à 3° suivant cet alinéa en vue de rendre cette obligation effective :
- l'information par le titulaire de l'autorisation de mise sur le marché (AMM), aux entreprises susceptibles d'assurer la reprise, de son intention de concéder l'exploitation ou de transférer l'autorisation du médicament ;
- la réponse motivée du titulaire de l'autorisation à chaque offre reçue ;
- la mise à disposition des entreprises candidates à la reprise, par le titulaire et, le cas échéant et sur la demande de celui-ci, par l'exploitant, de toutes les informations qui leur seraient nécessaires. Cette obligation exclut les informations dont la communication pourrait porter atteinte aux intérêts de l'entreprise. En outre, une obligation de confidentialité est prévue pour les entreprises candidates.
Aux termes du sixième alinéa du II, le titulaire est tenu d'informer l'ANSM dès qu'il envisage de retenir une offre de reprise et, hors de ce cas, au plus tard neuf mois après la notification de l'ANSM ayant généré l'obligation de recherche. Dans le rapport qu'il adresse alors à l'agence, le titulaire doit présenter les actions qu'il a engagées pour rechercher un repreneur, les offres reçues et justifier les réponses qu'il y a apportées. Enfin, le titulaire doit analyser dans son rapport la capacité d'approvisionnement de l'entreprise candidate.
B. Faute de repreneur et d'alternative disponible, une possible concession d'exploitation à un établissement pharmaceutique public
Le dernier volet d'obligations portant sur le titulaire de l'AMM est inscrit au dernier alinéa du nouveau II et intervient en cas d'absence de repreneur à la remise du rapport ou au terme du délai de neuf mois suivant la notification par l'ANSM.
Dans le cas où le besoin ne pourrait être couvert de manière pérenne et à la demande de l'ANSM, le titulaire de l'AMM serait tenu de concéder à titre gracieux l'exploitation et la fabrication du médicament pour le marché français.
Cette concession, d'une durée de deux ans reconductible, ne peut être faite qu'à un établissement pharmaceutique détenu par une personne publique. L'établissement pharmaceutique de l'Assistance publique - Hôpitaux de Paris (AP-HP), l'Agence générale des équipements et produits de santé (Ageps), est, selon la direction générale de la santé1061(*) « historiquement, et encore à ce jour, la seule structure publique en capacité de reprendre, à la demande des pouvoirs publics, des productions abandonnées par les industriels ».
L'établissement pharmaceutique de l'AP-HP dispose actuellement d'une ligne de production de formes injectables avec une remplisseuse automatique d'ampoules (jusqu'à 8 000 unités par lot), d'une ligne de production de gélules (jusqu'à 60 000 unités par lot) et d'une ligne de production de comprimés (jusqu'à 200 000 comprimés par lot) qui permettent d'assurer actuellement la production de la moitié de ses 40 références historiques.
L'autre moitié de la production a pu être maintenue grâce à une démarche de diversification des capacités de fabrication et d'exploitation de médicaments. Afin de sécuriser son activité, l'établissement pharmaceutique de l'AP-HP a fait le choix de transférer en sous-traitance l'ensemble de ses activités de production et de contrôle qualité (il reste propriétaire de ses fabrications et responsable de leur libération et mise sur le marché). L'externalisation totale de ses fabrications est prévue pour mi-2024.
Source : Réponses de l'Assistance publique - Hôpitaux de Paris au questionnaire de la rapporteure
Pour assurer l'exploitation et la fabrication, l'établissement pharmaceutique public désigné se verrait transmettre par l'ANSM les informations contenues dans le dossier d'AMM.
Enfin, la concession pourrait être cessée à l'initiative de l'ANSM si une entreprise mettait sur le marché français le même médicament ou un médicament similaire en capacité de couvrir de manière pérenne le besoin.
Le B de l'article prévoit la détermination, par décret en Conseil d'État, des modalités d'application du nouveau II.
C. Une obligation assortie de sanctions possibles
Le C prévoit le régime de sanctions applicables dans le cas où le titulaire d'une autorisation de mise sur le marché ou une entreprise exploitante ne mettrait pas en oeuvre les obligations prévues au nouveau II.
Il modifie à cette fin l'article L. 5124-18 du CSP qui prévoit les manquements soumis à sanction financière, laquelle, aux termes de l'article L. 5471-1, serait prononcée par l'ANSM à l'encontre de l'auteur du manquement. La pénalité financière peut ainsi atteindre 30 % du chiffre d'affaires réalisé lors du dernier exercice clos pour le produit ou le groupe de produits concernés, dans la limite d'un million d'euros.
Le D prévoit l'exclusion, pour cette sanction, de l'astreinte journalière prévue pour chaque jour de rupture d'approvisionnement constaté, qui n'est pas opérante sur ces dispositions.
Le E affecte à la Caisse nationale de l'assurance maladie le produit de la sanction, par dérogation à l'article L. 5312-4-1 du CSP.
D. Une possibilité de complément par l'ANSM de la liste des MITM faisant l'objet d'un plan de gestion des pénuries
L'article L. 5121-31 du code de la santé publique prévoit que pour les médicaments d'intérêt thérapeutique majeur, « les titulaires d'autorisation de mise sur le marché et les entreprises pharmaceutiques exploitant des médicaments élaborent et mettent en oeuvre des plans de gestion des pénuries dont l'objet est, dans l'intérêt des patients, de prévenir et de pallier toute rupture de stock ».
Aux termes du troisième alinéa de cet article, la liste des médicaments faisant l'objet d'un plan de gestion des pénuries est transmise à l'ANSM par les titulaires d'autorisation et les entreprises exploitantes.
Le F de l'article 36 propose de modifier ce troisième alinéa afin de prévoir que l'agence pourrait compléter cette liste si un MITM n'y figure pas. Cette inscription à l'initiative de l'ANSM serait faite après une procédure contradictoire.
Cette mesure s'appuie notamment sur une préconisation de la Cour des comptes1062(*), laquelle recommandait ainsi de « préciser la définition des médicaments d'intérêt thérapeutique majeur (MITM), en donnant à l'ANSM la compétence d'y inclure des médicaments qui le justifieraient, bien que non proposés par les industriels (et inversement d'en exclure) ».
II - Les modifications considérées comme adoptées par l'Assemblée nationale
Dans le texte sur lequel la Première ministre a engagé la responsabilité de son gouvernement en application de l'article 49, alinéa 3, de la Constitution, vingt amendements de la rapporteure générale Stéphanie Rist ont été retenus, tous rédactionnels.
Cet article est considéré comme ayant été adopté par l'Assemblée nationale, ainsi modifié.
III - La position de la commission
A. Un mécanisme bienvenu dans un contexte de tensions récurrentes, qui devra cependant être précisé
1. Un dispositif soutenu par la commission
La commission accueille favorablement le dispositif proposé, en cela qu'il tente d'apporter une réponse supplémentaire dans la prévention des arrêts ou difficultés d'approvisionnement pour certains médicaments d'intérêt thérapeutique majeur. En outre, la direction générale de la santé1063(*) a indiqué qu' « une attention toute particulière sera portée sur la couverture des besoins en pédiatrie et sur les maladies rares ».
Interrogée par la rapporteure1064(*), l'Agence nationale de sécurité du médicament et des produits de santé a indiqué que 426 arrêts de commercialisation, tous médicaments confondus, lui ont été déclarés en 2022. Cependant, elle précise que les MITM représentent 40 % de ce chiffre, quand seul l'arrêt de commercialisation de quelques spécialités est jugé potentiellement très problématique.
Par ailleurs, la rapporteure note que l'ANSM voit son rôle renforcé dans les différentes réponses aux problématiques de pénuries. Elle constate que l'agence s'estime en outre en capacité, au moyen des outils dont elle dispose, d'évaluer effectivement la couverture des besoins des patients français en cas de déclaration, mais aussi compétente pour mettre à disposition les informations contenues dans le dossier d'AMM.
Enfin, sur un plan juridique, la rapporteure constate que le dispositif retenu semble se conformer aux limites énoncées par le Conseil constitutionnel dans sa décision de 20141065(*) sur la loi « Florange ». Le Conseil avait alors notamment admis une obligation encadrée de mise à disposition d'informations en veillant également à la protection de la liberté de propriété et de la liberté d'entreprendre, ainsi qu'à la proportionnalité des sanctions.
2. Une mission de l'établissement pharmaceutique de l'AP-HP à préciser et valoriser
Concernant les possibilités de reprises par un établissement pharmaceutique public, la rapporteure constate, derrière le pluriel utilisé, qu'un seul établissement est aujourd'hui calibré pour cette mission.
L'AP-HP a d'ailleurs souligné que son établissement pharmaceutique, héritier de la Pharmacie centrale des hôpitaux de Paris, est le seul établissement pharmaceutique hospitalier civil public et, en pratique le seul opérateur public en capacité de remplir les missions de reprise d'AMM, quel que soit le cadre de la reprise.
Cependant, il convient de souligner que l'État vient aujourd'hui confier une nouvelle mission à l'Ageps après un virage stratégique important opéré depuis plusieurs années, décrit encore récemment dans les travaux de la commission d'enquête sénatoriale sur les pénuries de médicaments1066(*). La commission d'enquête, qui appelait à « arrêter le déclin de l'établissement pharmaceutique de l'Ageps », préconisait, au regard des enjeux actuels, de « restaurer la capacité de façonnage » de l'établissement.
Comme l'a précisé l'AP-HP, la reprise par l'Ageps de médicaments délaissés par l'industrie est envisageable selon deux modalités : soit reprendre en son nom l'exploitation des AMM concernées, avec une production en sous-traitance ; soit confier à un exploitant tiers le soin de fabriquer le produit.
Au titre des modalités opérationnelles, l'AP-HP a souligné qu'il « conviendrait de prévoir le financement des éventuelles productions non utilisées et de préciser le circuit de distribution de ces productions. Selon la nature et le volume des produits concernés, une distribution soit directement par l'EP de l'AP-HP, soit par Santé publique France, soit par un dépositaire ».
Enfin, alors que le texte ne mentionne pas directement le seul établissement public en capacité d'assurer la mission donnée, l'AP-HP s'est interrogée sur « l'élargissement de la possibilité de reprise d'exploitation d'AMM à l'ensemble “des établissements pharmaceutiques détenus par une personne morale de droit public” », estimant « nécessaire de regrouper les moyens autour de l'expertise existante, plutôt que de créer de nouvelles missions ou de nouveaux opérateurs et disperser ces moyens » et considérant ainsi que les pharmacies des centres hospitaliers universitaires doivent se fédérer autour d'un réseau appuyé sur un acteur hospitalier unique.
Enfin, la rapporteure souligne que le financement de cette mission nouvelle donnée à l'établissement pharmaceutique de l'AP-HP devrait être assuré, vraisemblablement par des dotations liées aux missions d'intérêt général.
La rapporteure s'interroge cependant sur les modalités de reprise effective et la capacité d'organisation rapide des acteurs dans un temps contraint dans le cas d'une absence de repreneur privé.
Sur ce point, la direction générale de la santé a indiqué que « dès la notification d'arrêt de commercialisation par un industriel, une information coordonnée de l'ANSM auprès de la DGS, l'EP de l'AP-HP et la direction générale des entreprises (DGE) est à établir afin de prendre le cas échéant le relais de la production des médicaments en évitant les ruptures de traitements pour les patients ».
La rapporteure souligne que la mobilisation éventuelle d'un établissement public dans le cas d'une lacune de reprise par une entreprise peut intervenir de manière tardive. En effet, l'article ne prévoit le cas de l'intervention de l'établissement pharmaceutique public qu'après la transmission du rapport requis de la part du titulaire ou de l'exploitant, lequel peut intervenir au bout de neuf mois, soit moins de trois mois avant l'arrêt de commercialisation.
Il paraît ainsi utile d'informer au plus tôt les établissements pharmaceutiques publics susceptibles de reprendre la production des décisions d'arrêts de commercialisation. Il s'agit ainsi de permettre à ces derniers d'engager une phase prospective dans de bonnes conditions et ainsi anticiper au mieux leur éventuelle reprise par concession de l'exploitation ou de la fabrication. C'est le sens de l'amendement n° 299 adopté par la commission.
Au-delà de l'établissement pharmaceutique désigné, la rapporteure s'est interrogée sur le rôle de l'agence nationale de santé publique, Santé publique France, dans le cadre de ce dispositif.
En effet, si Santé publique France n'est pas un exploitant, l'agence peut, aux termes de l'article L. 1413-4 du code de la santé publique, à la demande du ministre chargé de la santé, procéder notamment à l'acquisition, la fabrication, au transport et à la distribution et l'exportation de médicaments répondant à des besoins de santé publique, thérapeutiques ou diagnostiques, non couverts par ailleurs, qui font l'objet notamment d'une rupture ou d'une cessation de commercialisation. Elle peut enfin être titulaire d'une licence d'office mentionnée à l'article L. 613-16 du code de la propriété intellectuelle. L'agence pourrait donc être un appui, sans que soit précisée effectivement sa mission.
L'action de l'agence semble limitée aux situations d'urgence. Cependant, comme l'avait souligné la commission d'enquête sénatoriale sur les pénuries de médicament, Santé publique France a su, durant la crise des curares, montrer sa compétence en matière de gestion des stocks et circuits de distribution.
B. Des réserves sur l'efficacité réelle du dispositif
Au-delà d'une intention de principe qui peut être soutenue, le dispositif comporte néanmoins différents aspects opérationnels qui devront être précisés.
1. Un dispositif décrit par les industriels comme complexe et cernant mal les contraintes internationales
Ainsi, en réponse aux questions adressées par la rapporteure, le Leem a signalé certaines limites du dispositif proposé, indiquant qu'une reprise d'exploitation d'un MITM n'est possible que sur certains types de médicaments potentiellement productibles par le public, interrogeant notamment la situation des médicaments biologiques.
En outre, le Leem identifiait une lacune quant aux protections règlementaires concernant la commercialisation de génériques ou de biosimilaires. En outre, le Leem souligne que « la propriété des droits d'une AMM est très rarement française mais plutôt un actif de la société à l'échelle européenne voire très souvent internationale » : la décision de sa cession n'est ainsi pas une décision française. Insistant encore sur les réalités internationales de la structuration du secteur pharmaceutique et le fait que le titulaire d'AMM soit « très rarement un laboratoire français », le Leem a également soulevé auprès de la rapporteure la question des relations entre l'exploitant et le titulaire, mettant en avant des nécessités contractuelles et une possible complexité des procédures. À ce titre, le Leem s'interroge d'ailleurs « sur la capacité d'une structure publique à remplir les obligations liées à l'exploitation d'AMM et à conserver les liens avec le titulaire d'AMM ».
D'un point de vue opérationnel, le Leem a également interpellé sur le délai de neuf mois prévu par le texte, qui lui apparaît court « dans le cadre des négociations contractuelles et d'une stratégie de décision au niveau international ». Ce délai semble cependant à la rapporteure difficile à allonger, alors que ce délai débute moins d'un an avant l'arrêt possible de la commercialisation du médicament.
En conclusion, le Leem, qui a insisté sur les incertitudes juridiques résultant d'une rédaction qu'il juge « peu claire et trop large », y voyant une éventuelle « entrave à la liberté d'entreprendre de l'entreprise », signale que « cette mesure, au-delà de la complexification toujours croissante de l'environnement règlementaire en France risque d'avoir des effets contre-productifs. En effet, la mise en place d'un tel dispositif risque de dissuader d'éventuels repreneurs de ces MITM de se manifester sur le territoire français ».
2. D'éventuelles conséquences non anticipées sur les prix
Enfin, le comité économique des produits de santé (CEPS) a signalé un point de vigilance sur cet article, considérant « le fait que cette obligation de rechercher un repreneur ou même le fait de confier la licence à un tiers, puisse faire naître des contraintes spécifiques en matière de prix qui ne sont pas anticipées dans la loi », estimant que « ce serait le cas si l'acceptabilité par un repreneur, fût-il public et désigné par l'État, était conditionnée par ce repreneur à une hausse de prix n'entrant pas nécessairement dans le cadre conventionnel ».
3. Des solutions complémentaires suggérées par l'ANSM
Par ailleurs, sur un autre plan, l'ANSM a également signalé à la rapporteure que la fabrication sous licence « pourrait être un moyen intéressant » pour permettre la poursuite de la fabrication de médicaments qui ne sont plus sous brevet et pour lesquels les laboratoires n'ont pas trouvé de repreneur, dans le cas où ceux-ci souhaitent néanmoins garder la titularité de leur AMM.
C. Un renforcement souhaité du pouvoir de l'ANSM en matière de plans de gestion des pénuries
La rapporteure souscrit à l'ouverture d'une possibilité donnée à l'ANSM d'intervenir plus efficacement sur la liste des MITM faisant l'objet d'un plan de gestion des pénuries. Cette modification, qui certes ferait l'objet d'une procédure contradictoire, sera in fine la décision de l'agence.
Cependant, cette disposition apparaît insuffisamment opérationnelle. C'est pourquoi la commission, à l'initiative de la rapporteure, a adopté un amendement n° 300 visant à donner la possibilité pour l'Agence nationale de sécurité du médicament et des produits de santé de désigner des médicaments d'intérêt thérapeutique majeur devant faire l'objet d'un plan de gestion des pénuries dans le cas où l'entreprise exploitante ne les aurait pas identifiés comme tels. Cette formulation traduit plus directement les recommandations de la Cour des comptes et suit également les recommandations de la commission d'enquête sénatoriale1067(*) qui appelait à « permettre à l'ANSM d'inclure ou d'exclure des spécialités » de la liste des MITM. Cet amendement préserve la procédure contradictoire proposée.
Il convient de noter qu'en prévoyant la modification de cette liste, la disposition satisfait l'objet de la disposition remplacée dans l'article 36 dans la mesure où un médicament qualifié de MITM devant faire l'objet d'un plan de gestion des pénuries.
En outre, le même amendement, suivant également les recommandations de la commission d'enquête, prévoit avec la publication par l'ANSM de la liste des MITM une utile mesure de transparence.
Enfin, alors que la commission d'enquête préconisait une priorisation des plans de gestion des pénuries, le présent amendement prévoit une catégorie de « plans de gestions renforcés » pour les MITM pour lesquels une rupture ou un risque de rupture de stock identifiés.
D. Des dispositions à la recevabilité sociale cependant discutable
Dans l'étude d'impact jointe au PLFSS, le Gouvernement justifie la place de la disposition en LFSS par un impact :
- en dépenses, en réduisant le coût des importations nécessaires à la couverture des besoins générés par l'arrêt de commercialisation d'un MITM ;
- en recettes, par l'affectation du produit de la pénalité à l'assurance maladie.
Or, l'effet financier décrit par le Gouvernement apparaît très indirect. En effet, selon des hypothèses particulièrement fragiles retenues dans l'étude d'impact, l'économie pourrait aller de 10 millions d'euros à 40 millions d'euros. Ce montant, qui ne relève que d'une économie éventuelle et entourée de beaucoup d'incertitudes, ne saurait être qualifié d'effet direct sur les comptes de l'assurance maladie.
En outre, la jurisprudence constatée en matière de recevabilité sociale tend à ne pas apprécier le produit d'une sanction comme un effet en recettes, le rendement étant par hypothèse attendu nul.
Partant, les dispositions du présent article tendent à devoir être regardées, comme d'autres mesures relatives aux ruptures d'approvisionnement portées dans de précédentes LFSS, comme relevant d'une politique de sécurité sanitaire et non comme une disposition ayant un effet direct sur les régimes de base de la sécurité sociale aux termes de la loi organique.
La commission estime donc qu'elles pourraient être considérées comme un « cavalier social » et qu'il existe un risque non négligeable de censure par le Conseil constitutionnel. Un projet de loi consacré aux dispositions relatives aux pénuries de médicament ou portant diverses dispositions d'ordre sanitaire et social aurait été un véhicule sans doute plus approprié.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article
36 bis (nouveau)
Sortie de l'expérimentation relative à
l'usage médical du cannabis et dispositif d'autorisation provisoire du
cannabis à usage médical
Cet article organise la sortie de l'expérimentation de l'usage médical du cannabis en créant une période transitoire jusqu'au 31 décembre 2024 et, dans l'attente d'une autorisation de mise sur le marché, crée une procédure d'autorisation provisoire pour le cannabis à usage médical.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - Le dispositif proposé
A. Une expérimentation initiée par la loi de financement de la sécurité sociale pour 2020 qui prendra fin le 26 mars 2024
L'expérimentation de l'usage médical du cannabis a été inscrite dans l'article 43 de la loi n° 2019-1446 du 24 décembre 2019 de financement de la sécurité sociale (LFSS) pour 20201068(*). Initiée en mars 2021 pour une durée de deux ans, elle a été prolongée d'une année par la LFSS pour 20231069(*) ; elle doit prendre fin le 26 mars 2024.
Les conditions de mise en oeuvre de cette expérimentation ont été définies par voie réglementaire. Ainsi, le décret n° 2020-1230 du 7 octobre 2020 relatif à l'expérimentation de l'usage médical du cannabis prévoit qu'elle porte sur un nombre maximal de 3 000 patients. Au 14 septembre 2023, selon le Gouvernement, 2 761 patients avaient été inclus dans l'expérimentation.
Le cannabis à usage médical peut être prescrit dans cinq indications limitatives fixées par arrêté1070(*) : les douleurs neuropathiques réfractaires aux thérapies accessibles, certaines formes d'épilepsie pharmaco-résistantes, certains symptômes rebelles en oncologie liés au cancer ou au traitement anti-cancéreux, les situations palliatives et la spasticité douloureuse de la sclérose en plaques ou d'autres pathologies du système nerveux central.
Au 13 décembre 2022, parmi les 2 296 patients inclus dans l'expérimentation, la répartition par indication était la suivante : 833 patients pour douleurs neuropathiques réfractaires ; 226 patients pour une spasticité douloureuse dans la sclérose en plaques ; 181 patients dans les épilepsies pharmaco résistantes ; 114 patients en situation palliative ; 111 patients en oncologie1071(*).
Au cours de la première phase de l'expérimentation, les médicaments utilisés par les patients inclus dans cette expérimentation étaient fournis à titre gratuit par les entreprises. Depuis le 27 mars 2023, il est prévu que « la dispensation et le renseignement du registre de suivi des patients [...] sont facturés quinze euros toutes taxes comprises à l'assurance maladie. [...] Une seule facturation par dispensation est autorisée quels que soit la forme, le conditionnement et le nombre d'unités délivrées. La dispensation bénéficie d'une prise en charge intégrale par l'assurance maladie obligatoire. »1072(*)
B. Une procédure d'autorisation provisoire du cannabis à usage médical hors autorisation de mise sur le marché
Le présent article est issu d'un amendement inséré par le Gouvernement dans le texte sur lequel il a engagé sa responsabilité devant l'Assemblée nationale en application du troisième alinéa de l'article 49 de la Constitution. Il vise à définir une procédure d'autorisation provisoire du cannabis à usage médical hors autorisation de mise sur le marché.
1. Une autorisation temporaire délivrée par l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) dans le cadre d'une procédure ad hoc
Le présent article propose tout d'abord la création d'un régime d'autorisation provisoire pour le cannabis à usage médical. À cette fin, il est proposé de rétablir le 4° de l'article L. 5121-1 du code de la santé publique1073(*).
Selon le premier alinéa de ce 4°, le médicament à base de cannabis est entendu comme « tout médicament dont la substance active est composée d'une préparation à base de Cannabis sativa L. », qui respecte les bonnes pratiques de fabrication1074(*) et les spécifications techniques portant sur les caractéristiques, la composition et la forme pharmaceutique du médicament, ces spécifications étant fixées par arrêté du ministre de la santé, sur proposition de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM).
Selon le deuxième alinéa du même 4°, l'utilisation du cannabis à usage médical est subordonnée à une autorisation temporaire de l'ANSM pour une durée maximale de cinq ans1075(*). L'autorisation vise des indications thérapeutiques et des situations cliniques particulières, définies par arrêté du ministre de la santé. Il s'agira de répondre aux besoins des patients en l'absence de spécialité pharmaceutique adaptée autorisée au titre de l'une des procédures suivantes : procédure nationale d'autorisation de mise sur le marché (AMM)1076(*) ; autorisation dérogatoire délivrée par l'ANSM pour des raisons de santé publique1077(*) ; autorisation au titre de l'accès précoce1078(*) ou de l'accès compassionnel1079(*) ; autorisation d'importation, notamment en cas de rupture d'un stock de médicament.
Le Gouvernement indique que l'accès au cannabis à usage médical sera restreint en dernière ligne de traitement, ouvert uniquement sur prescription hospitalière initiale, pour des indications et situations cliniques « pour lesquelles l'efficacité pourrait être présumée »1080(*). Il ne précise pas si les indications actuellement prévues par arrêté dans le cadre de l'expérimentation en cours seraient reconduites.
Un article L. 5121-15 serait également rétabli dans le code de la santé publique afin de créer un nouveau régime dérogatoire d'autorisation par l'ANSM des médicaments à base de cannabis mentionnés au 4° de l'article L. 5121-1 précité. L'utilisation de ces médicaments serait autorisée pour une durée temporaire limitée à cinq ans, l'autorisation étant renouvelable par période de cinq ans. Des conditions particulières accompagnent la délivrance de cette autorisation au demandeur :
- celui-ci est nécessairement établi dans un État membre de l'Union européenne ou de l'Espace économique européen ;
- il a l'obligation de recueillir les données de suivi des patients traités et de transmettre chaque année une synthèse de ces données à l'ANSM ;
- la réalisation d'études de sécurité ou d'efficacité après délivrance de l'autorisation peut également être exigée.
L'article L. 5121-15 précise en outre les conditions dans lesquelles la délivrance d'une autorisation pourrait être refusée. Tel serait le cas, en particulier, lorsque l'indication thérapeutique proposée ne serait pas autorisée, l'évaluation de la balance bénéfices-risques ne serait pas favorable ou la composition du médicament ne serait pas conforme à ce que déclare le demandeur.
L'article L. 5121-15 précise enfin les cas dans lesquels l'autorisation peut être suspendue, retirée ou modifiée1081(*). Chacune de ces décisions fait l'objet d'une publicité immédiate la plus large possible à destination des patients et des professionnels de santé.
En outre, un article L. 5121-14-2-1 serait créé dans le code de la santé publique afin de compléter les procédures de modification, suspension ou retrait de d'autorisation prévues à l'article L. 5121-15. Aux termes de cet article, l'ANSM serait autorisée, en dehors des procédures précitées, à retirer du marché les médicaments à base de cannabis dans l'intérêt de la santé publique, notamment si le médicament n'a pas la composition déclarée ou si l'une des exigences qui conditionne l'octroi de l'autorisation n'a pas été respectée1082(*). En cas de retrait de l'autorisation, l'ANSM pourrait néanmoins, dans des circonstances exceptionnelles et pour une durée provisoire, autoriser la délivrance du médicament à des patients déjà traités avec lui.
2. Des dispositions complémentaires pour prévenir des dérives éventuelles liées à l'autorisation du cannabis à usage médical
Le présent article propose d'instaurer diverses mesures d'encadrement destinées à éviter de possibles dérives.
Il s'agit en premier lieu de prévenir tout risque de publicité des médicaments à base de cannabis par le titulaire de l'autorisation temporaire. L'article L. 5121-16 du code la santé publique est rétabli à cette fin.
L'article L. 5421-6-3 du code de la santé publique, également rétabli, prévoirait les sanctions qui punissent :
- la fabrication, la commercialisation ou la distribution des médicaments à base de cannabis en dehors de toute autorisation de l'ANSM (I) ;
- le fait de contribuer à toute mesure de publicité, directement ou indirectement, sur un médicament à base de cannabis, ou la diffusion d'informations aux professionnels de santé en dehors du cadre fixé par la décision de l'ANSM (II).
Ces sanctions, fixées à cinq ans d'emprisonnement et 375 000 euros d'amende, seraient relevées à sept ans d'emprisonnement et 750 000 euros d'amende lorsque l'une des circonstances précisées au III du même article est remplie - risque grave pour la santé de l'homme, commission en bande organisée, commission sur un réseau de télécommunication à destination d'un public non déterminé ou commission par certains professionnels énumérés.
L'article L. 5422-18 du code de la santé publique, qui énumère divers manquements soumis à sanction financière en matière de publicité de médicaments, serait complété d'un nouvel alinéa (20°) qui vise à y inscrire toute diffusion d'information au public relative à l'autorisation d'un médicament à base de cannabis ou la diffusion de l'information auprès des professionnels de santé en dehors du cadre fixé par l'ANSM.
L'article L. 5121-20 du code de la santé publique serait complété d'un 20° afin de déterminer divers éléments qui devront faire l'objet de précisions par un décret en Conseil d'État. Il s'agit notamment des modalités de présentation des demandes d'autorisation temporaire des médicaments à base de cannabis, des conditions dans lesquelles interviennent les décisions de l'ANSM relatives à l'octroi, au refus, à la modification, au renouvellement, à la suspension et au retrait des autorisations temporaires délivrées, ainsi que des règles applicables en cas de changement du titulaire de l'autorisation.
Le code de la santé publique serait enfin modifié par diverses dispositions de cohérence rédactionnelle (2°, 4°, 8°, 9° et 10° du I du présent article).
3. Des conditions de prise en charge par l'assurance maladie subordonnées à un avis préalable de la HAS
Aux termes du nouvel article L. 162-17-2-4 du code de la sécurité sociale, les règles de prise en charge par l'assurance maladie des médicaments à base de cannabis autorisés par l'ANSM seraient définies par décret en Conseil d'État.
La Haute Autorité de santé (HAS) serait chargée de l'évaluation de ces médicaments, en particulier des indications médicales qui justifient leur usage, ainsi que des conditions dans lesquelles ces médicaments peuvent être pris en charge par l'assurance maladie. Les conditions de cette prise en charge seraient définies par arrêté du ministre de la santé, de même que le prix de vente du médicament par le fabricant aux officines ou établissements de santé. En cas d'avis défavorable de la HAS, ces médicaments ne seraient pas pris en charge par l'assurance maladie.
Le prix de vente tiendrait compte de diverses caractéristiques du médicament, ainsi que « des prix ou des tarifs européens présentant une taille totale de marché comparable déterminés par décret. Ce prix ne peut être supérieur aux prix fixés pour une spécialité comparable ou à même visée thérapeutique. »
4. Une période transitoire pour assurer la continuité de prise en charge des patients inclus dans l'expérimentation
L'article 43 de la loi du 24 décembre 2019 de financement de la sécurité sociale pour 2020, qui a fixé le cadre juridique de l'expérimentation, serait quant à lui modifié pour :
- substituer à son terme actuel, soit le 26 mars 2024, une nouvelle échéance correspondant à celle de la disponibilité d'un médicament à base de cannabis autorisé par l'ANSM, au plus tard le 31 décembre 2024 ;
- prévoir que les modalités de prise en charge de ces médicaments par l'assurance maladie sont définies par arrêté du ministre de la santé dans les conditions prévues à l'article L. 162-17-2-4 précité, à partir du 26 mars 2024 et jusqu'au 31 décembre 2024 au plus tard ;
- supprimer l'alinéa (III) qui prévoit qu'un rapport est remis par le Gouvernement au Parlement six mois avant son terme, réalisant un bilan de l'expérimentation, la pertinence de l'élargissement de l'usage du cannabis thérapeutique et sur les modalités de sa prise en charge par l'assurance maladie.
L'instauration d'une période transitoire permet d'assurer une continuité de prise en charge des patients inclus dans l'expérimentation jusqu'au 26 mars 2024. La procédure d'autorisation temporaire du cannabis à usage médical aurait ensuite vocation à pérenniser l'usage médical du cannabis. Elle nécessite la publication de textes réglementaires qui pourraient intervenir au cours de la période transitoire.
Seuls les patients inclus dans l'expérimentation pourront continuer à utiliser des médicaments à base de cannabis au cours de cette période transitoire. Ces médicaments seront pris en charge par l'assurance maladie sur la base d'un montant fixé par arrêté ministériel. Le Gouvernement évalue le coût de la période transitoire à 10 millions d'euros.
II - La position de la commission
Après avoir constaté que l'expérimentation de l'usage médical du cannabis a débuté il y a près de trois ans, la commission a rappelé que la loi prévoit actuellement qu'elle doit faire l'objet d'un rapport du Gouvernement au Parlement dans un délai de six mois avant son terme. Ce rapport doit permettre d'évaluer le résultat de l'expérimentation, de statuer sur l'opportunité d'un élargissement des indications autorisées et sur les conditions de prise en charge par l'assurance maladie des médicaments à base de cannabis.
À rebours d'une décision sereine qui supposerait de pouvoir s'appuyer sur un bilan formalisé et d'aménager un temps de réflexion pour envisager les suites à réserver à cette expérimentation, le Gouvernement fait preuve de précipitation en inscrivant dès à présent dans la loi des dispositions qui pérennisent le recours au cannabis à usage médical. Plus que cela, il est regrettable que le Gouvernement, en supprimant la disposition qui prévoit la remise d'un rapport au Parlement, cherche ainsi à s'affranchir d'une évaluation de l'expérimentation que commande pourtant le sens des responsabilités ou, à défaut, le seul bon sens, dans une matière qui n'engage pas moins que la sécurité des patients.
De l'aveu du Gouvernement lui-même, aucune preuve clinique ne permet aujourd'hui de satisfaire aux prérequis d'une autorisation de mise sur le marché pour ces médicaments. La commission ne peut donc admettre que le principe de l'évaluation soit ainsi supprimé, comme un accessoire inutile et encombrant, privant la représentation nationale de son droit de regard.
En outre, l'ANSM signalait en mars dernier un risque de rupture d'approvisionnement sur les médicaments à base de cannabis1083(*). Elle recommandait alors, afin de garantir la poursuite du traitement des patients inclus dans l'expérimentation pour lesquels aucune autre alternative n'existe, de suspendre les inclusions de patients nécessitant un traitement par des médicaments à base de cannadibiol seul (CBD 50 LGP Classic). La commission souhaite être éclairée sur ce risque de rupture, les actions entreprises pour y remédier et l'état de la situation actuelle.
Dans ce contexte, à l'initiative de sa rapporteure, la commission propose :
- par l'amendement n° 301, de réintroduire l'évaluation de l'expérimentation de l'usage médical du cannabis, qui devra faire l'objet d'un rapport au Parlement au plus tard le 30 juin 2024 ;
- par l'amendement n° 302, de solliciter pour avis la HAS sur les modalités de prise en charge par l'assurance maladie des médicaments à base de cannabis en fonction des indications thérapeutiques autorisées, au regard des résultats de l'expérimentation.
Sur la base de ces informations, le législateur pourra, le cas échéant, adapter les dispositions autorisant la pérennisation de l'usage médical du cannabis.
L'aménagement d'une période transitoire jusqu'au 31 décembre 2024, au bénéfice des seuls patients inclus, apparaît en revanche adapté.
La commission propose d'adopter cet article modifié par les amendements qu'elle a adoptés.
* 937 Rapport d'activité 2022 de l'EFS.
* 938 L'adjectif « labile » signifie « qui est sujet à changer, à se transformer ». Dans le contexte de la filière sang, il désigne les produits sanguins destinés à être transfusés.
* 939 La société Octapharma est aujourd'hui le seul concurrent de l'EFS sur le marché français.
* 940 Cour des comptes, La filière du sang en France : un modèle économique fragilisé, une exigence de transformation, rapport public annuel 2019, février 2019.
* 941 En moyenne depuis 2012, les recettes de l'EFS sont d'environ 1 milliard d'euros, dont environ 870 millions d'euros proviennent de la cession de produits sanguins labiles.
* 942 Lors de son audition, l'EFS a indiqué qu'une diminution de cession des concentrés de globules rouge de 1% représente une perte de chiffre d'affaires de l'ordre de 5 millions d'euros. Ainsi, en 2022, la baisse enregistrée de 4,6 % a généré un déficit de recettes estimé à plus de 22 millions d'euros.
* 943 La Cour de justice de l'Union européenne a jugé (CJUE 5 octobre 2016, n° 412/15) que la livraison de sang humain, y compris la livraison du plasma qui entre dans sa composition, pouvait bénéficier de l'exonération de TVA lorsque cette livraison contribuait directement à des activités d'intérêt général, à savoir lorsque le plasma livré était directement employé pour des soins de santé ou à des fins thérapeutiques. Selon la réponse de la ministre des solidarités et de la santé au chapitre précité du rapport public annuel 2019 de la Cour des comptes, « l'impact pour l'EFS est une perte de 76,5 millions d'euros par an : 52,6 millions d'euros liés à une moindre récupération de TVA plus 23,9 millions d'euros liés à une augmentation de la taxe sur les salaires ».
* 944 Source : Annexe 9 du présent PLFSS.
* 945 L'EFS a été présidé du 15 octobre 2012 au 15 octobre 2023 par François Toujas, ensuite nommé à la présidence de l'Office national d'indemnisation des accidents médicaux (Oniam) ; depuis le 16 octobre 2023, Pascal Morel assure la présidence par intérim.
* 946 « Les frais occasionnés par le prélèvement et le conditionnement des produits et organes d'origine humaine sont remboursés par les caisses lorsqu'un tarif de responsabilité a été fixé par arrêté interministériel ».
* 947 Le présent article se réfère au « 1° de l'article L. 1222-8 du code de la santé publique », relatif aux « produits des activités liées aux produits sanguins labiles ».
* 948 Fiches d'évaluation préalable.
* 949 L'annexe 9 indique que cette dotation sera divisée en sous-enveloppes, par exemple celle dédiée à l'activité d'immunohématologie et à la délivrance déficitaire, celle dédiée à la recherche ou encore aux thérapies innovantes.
* 950 Une disposition identique figure à l'article L. 756-2-1 du code de l'éducation, relatif au financement de l'École des hautes études en santé publique (« la participation des organismes d'assurance maladie est versée et répartie entre les régimes dans des conditions fixées par décret »).
* 951 Élisabeth Doineau, Annie Le Houerou, Dotations de la sécurité sociale : sortir de la logique du financement à l'aveugle, rapport d'information n° 877 (2022-2023), mission d'évaluation et de contrôle de la sécurité sociale (Mecss) de la commission des affaires sociales du Sénat, 12 juillet 2023.
* 952 Voir, à ce sujet, le commentaire de l'article 33 du présent PLFSS.
* 953 Rapport n° 828 (2022-2023) « Pénurie de médicaments : Trouver d'urgence le bon remède », tome I, déposé le 4 juillet 2023.
* 954 Ibid., pp. 83 à 85 : entre 2012 et 2022, la consommation mondiale de médicaments a augmenté de plus de 36 %.
* 955 Ibid., pp. 76 à 83.
* 956 Ibid., p. 88.
* 957 Ibid., pp. 100 à 109.
* 958 Article 151 de la loi n° 2016-41 du 26 janvier 2016 de modernisation de notre système de santé.
* 959 Article 48 de la loi n° 2019-1446 du 24 décembre 2019 de financement de la sécurité sociale pour 2020.
* 960 Article L. 5121-32 du code de la santé publique.
* 961 Les PUI des centres hospitaliers de Lille, de Lyon, de Nîmes, de Rennes, de Bordeaux et de Toulouse ont été mobilisées.
* 962 Annexe n° 9 « Fiches d'évaluation préalable des articles du projet de loi » jointe au PLFSS pour 2022, p. 280.
* 963 Article 61 de la loi n° 2021-1754 du 23 décembre 2021 de financement de la sécurité sociale pour 2022.
* 964 Article L. 5126-6 du code de la santé publique.
* 965 Article L. 5121-1 du code de la santé publique.
* 966 Voir, notamment, le rapport précité de la commission d'enquête sénatoriale relative aux pénuries de médicament, ou le débat annuel sur l'application des lois de 2023.
* 967 Article L. 5121-1 du code de la santé publique.
* 968 Article L. 5125-1 du code de la santé publique.
* 969 Article L. 5121-5 du code de la santé publique.
* 970 Article R. 5125-9 du code de la santé publique.
* 971 Article L. 5125-1-1-1 du code de la santé publique.
* 972 Article L. 5125-1-1 du code de la santé publique.
* 973 Arrêté du 14 novembre 2014 fixant la liste des préparations pouvant présenter un risque pour la santé mentionnées à l'article L. 5125-1-1 du code de la santé publique.
* 974 Recommandation du 29 décembre 2023 de la directrice générale de l'ANSM établie en application du V. de l'article L. 5125-23 du code de la santé publique.
* 975 Voir, sur ce point, le rapport de la commission d'enquête sénatoriale précité, p. 210.
* 976 Audition de Mme Christelle Ratignier-Carbonneil, directrice générale de l'Agence nationale de sécurité du médicament et des produits de santé (ANSM), par la commission d'enquête sénatoriale sur les pénuries de médicaments le 15 février 2023.
* 977 Voir le rapport n° 130, tome I (2021-2022) de Mmes Élisabeth Doineau, rapporteure générale, Corinne Imbert, MM. René-Paul Savary, Olivier Henno, Mme Pascale Gruny et M. Philippe Mouiller, déposé le 3 novembre 2021, pp. 292-293.
* 978 Réponses écrites de l'AP-HP au questionnaire transmis par la rapporteure.
* 979 Audition de M. François Braun, ministre de la santé et de la prévention par la commission d'enquête sénatoriale sur la pénurie de médicaments et les choix de l'industrie pharmaceutique française, le 15 juin 2023.
* 980 Amendement n° 176 au PLFSS pour 2022 présenté par Mme Corinne Imbert, au nom de la commission des affaires sociales, et adopté par le Sénat.
* 981 Rapport n° 828 (2022-2023) « Pénurie de médicaments : Trouver d'urgence le bon remède » précité, recommandation n° 15.
* 982 Article L. 5138-1 du code de la santé publique.
* 983 Article L. 5138-5 du code de la santé publique.
* 984 Décret n° 2012-1096 du 28 septembre 2012 relatif à l'approvisionnement en médicaments à usage humain.
* 985 Article R. 5124-49-1 du code de la sécurité sociale.
* 986 Article 151 de la loi n° 2016-41 du 26 janvier 2016 de modernisation de notre système de santé.
* 987 Article 48 de la loi n° 2019-1446 du 24 décembre 2019 de financement de la sécurité sociale pour 2020.
* 988 Article L. 5121-32 du code de la santé publique.
* 989 Les causes déclarées de ces ruptures sont présentées dans le commentaire de l'article 32 du présent PLFSS.
* 990 Ces données sont disponibles sur la plateforme de consultation mise à disposition par l'ANSM : https://data.ansm.sante.fr/ruptures.
* 991 Rapport n° 828 (2022-2023) « Pénurie de médicaments : Trouver d'urgence le bon remède », tome I, déposé le 4 juillet 2023, p. 105.
* 992 Article 151 de la loi n° 2016-41 du 26 janvier 2016 de modernisation de notre système de santé.
* 993 Article L. 5111-4 du code de la santé publique.
* 994 Article 48 de la loi n° 2019-1446 du 24 décembre 2019 de financement de la sécurité sociale pour 2020.
* 995 Article L. 5121-31 du code de la santé publique.
* 996 Article R. 5124-49-5 du code de la santé publique.
* 997 Article R. 5124-49-1 du code de la santé publique.
* 998 Ibid.
* 999 Article L. 5121-32 du code de la santé publique.
* 1000 Article 48 de la loi n° 2018-1203 du 22 décembre 2018 de financement de la sécurité sociale pour 2019.
* 1001 Article R. 5124-49-4 du code de la santé publique.
* 1002 Article 47 de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
* 1003 Décret n° 2012-1096 du 28 septembre 2012 relatif à l'approvisionnement en médicaments à usage humain.
* 1004 Article R. 5124-59 du code de la santé publique.
* 1005 Article L. 5121-30 du code de la santé publique.
* 1006 Article L. 5124-17-3 du code de la santé publique.
* 1007 Rapport n° 828 (2022-2023) « Pénurie de médicaments : Trouver d'urgence le bon remède », op. cit., pp. 115 et 127 à 129.
* 1008 Article L. 5121-9 du code de la santé publique.
* 1009 Rapport n° 828 (2022-2023) « Pénurie de médicaments : Trouver d'urgence le bon remède », op. cit., pp. 116 à 118.
* 1010 Arrêté du 1er août 2016 déterminant la liste des tests, recueils et traitements de signaux biologiques qui ne constituent pas un examen de biologie médicale, les catégories de personnes pouvant les réaliser et les conditions de réalisation de certains de ces tests, recueils et traitements de signaux biologiques modifié.
* 1011 Article L. 5121-12-1-1 du code de la santé publique.
* 1012 Ces éléments sont plus précisément décrits dans le commentaire de l'article 25 du présent PLFSS.
* 1013 Stratégie nationale 2022-2025 de prévention des infections et de l'antibiorésistance, Santé humaine, action n° 21.
* 1014 Article L. 5121-33 du code de la santé publique.
* 1015 Article L. 5121-12-1-1 du code de la santé publique.
* 1016 Article L. 5123-8 du code de la santé publique.
* 1017 Soit 1 601 déclarations ayant donné lieu à au moins une mesure sur 3 761 déclarations reçues. Ces données sont disponibles sur la plateforme précitée : https://data.ansm.sante.fr/ruptures.
* 1018 Rapport n° 828 (2022-2023) « Pénurie de médicaments : Trouver d'urgence le bon remède », op. cit., recommandation n° 24.
* 1019 Cnam, Améliorer la qualité du système de santé et maîtriser les dépenses. Propositions de l'assurance maladie pour 2024, p. 208.
* 1020 Voir, à ce sujet, le commentaire de l'article 25 du présent PLFSS.
* 1021 Rapport n° 828 (2022-2023) « Pénurie de médicaments : Trouver d'urgence le bon remède », op. cit., p. 60.
* 1022 Ibid., pp. 123 et suivantes.
* 1023 Laquelle regroupe les actes des chirurgiens-dentistes, des sages-femmes et des auxiliaires médicaux.
* 1024 Article L. 165-1-5 du code de la sécurité sociale.
* 1025 Annexe 9 au PLFSS, p. 331.
* 1026 Idem.
* 1027 Règlement (CE) n° 1394/2007 du Parlement européen et du Conseil du 13 novembre 2007 concernant les médicaments de thérapie innovante et modifiant la directive 2001/83/CE ainsi que le règlement (CE) n° 726/2004.
* 1028 Article L. 162-22-7 du code de la sécurité sociale.
* 1029 Article 54 de la loi n° 2022-1616 du 23 décembre 2022 de financement de la sécurité sociale pour 2023.
* 1030 Article L. 162-16-6 du code de la sécurité sociale.
* 1031 Ibid.
* 1032 Exposé sommaire de l'amendement n° 3086 au PLFSS 2024, retenu dans le texte sur lequel le Gouvernement a engagé sa responsabilité.
* 1033 Rapport n° 99, tome II (2022-2023) de Mmes Élisabeth Doineau, rapporteure générale, Corinne Imbert, Pascale Gruny, MM. René-Paul Savary, Olivier Henno et Philippe Mouiller, déposé le 2 novembre 2022.
* 1034 En droit national, l'article L. 5121-9 du code de la santé publique dispose que l'AMM est refusée « lorsqu'il apparaît que l'évaluation des effets thérapeutiques positifs du médicament ou produit au regard des risques pour la santé du patient ou la santé publique liés à sa qualité, à sa sécurité ou à son efficacité n'est pas considérée comme favorable, ou qu'il n'a pas la composition qualitative et quantitative déclarée, ou que l'effet thérapeutique annoncé fait défaut ou est insuffisamment démontré par le demandeur. Elle est également refusée lorsque la documentation et les renseignements fournis ne sont pas conformes au dossier qui doit être présenté à l'appui de la demande. »
* 1035 Règlement (CE) n° 726/2004 du Parlement européen et du Conseil, du 31 mars 2004, établissant des procédures communautaires pour l'autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments
* 1036 Article L. 5121-9-1 du code de la santé publique.
* 1037 Article L. 5121-8 du code de la santé publique.
* 1038 Article L. 5121-9 du même code.
* 1039 Article L. 5121-8-1 dudit code.
* 1040 Article L. 5121-9-4 du même code : cette obligation concerne l'ensemble des actions engagées dans l'Union européenne, et les actions fondées sur un motif pouvant justifier le retrait de l'AMM engagées dans les pays tiers.
* 1041 1° de l'article L. 161-37 du code de la sécurité sociale.
* 1042 Article R. 163-3 du code de la sécurité sociale.
* 1043 Article L. 162-16-4-3 du code de la sécurité sociale.
* 1044 Article L. 162-22-7 et L. 162-23-6 du code de la sécurité sociale.
* 1045 Article L. 5126-6 du code de la santé publique.
* 1046 Ces chiffres ont été obtenus entre 2017 et 2020.
* 1047 Article 78 de la loi n° 2020-1576 du 14 décembre 2020 de financement de la sécurité sociale pour 2021.
* 1048 III de l'article L. 5121-12 du code de la santé publique.
* 1049 Article D. 5121-69-3 du code de la santé publique.
* 1050 Ce délai peut être fixé à quatre mois en cas de forte demande. Passé ce délai, le silence de la HAS vaut autorisation d'accès précoce.
* 1051 Article D. 5121-69-3 du code de la santé publique.
* 1052 III de l'article L. 5121-12 du code de la santé publique.
* 1053 Article L. 162-16-5-1 du code de la sécurité sociale.
* 1054 Article L. 162-22-7-3 du code de la sécurité sociale.
* 1055 En cas d'inscription au remboursement. En cas d'inscription sur la seule liste des médicaments agréés à l'usage des collectivités, le CEPS fixe un prix de référence.
* 1056 Article L. 5121-12-1 du code de la santé publique.
* 1057 Article L. 162-22-7-3 du code de la sécurité sociale.
* 1058 Leur liste sera définie par décret.
* 1059 Loi n° 2014-384 du 29 mars 2014 visant à reconquérir l'économie réelle.
* 1060 Ce cas couvre, comme l'explique l'étude d'impact, par exemple, « le cas où le médicament en question n'est pas la seule offre possible, mais qu'il y a déjà une forte tendance de diminution de l'offre ».
* 1061 Réponses au questionnaire de la rapporteure.
* 1062 Rapport public annuel 2022.
* 1063 Réponses au questionnaire adressé.
* 1064 Réponses au questionnaire adressé.
* 1065 Décision n° 2014-692 DC du 27 mars 2014.
* 1066 Pénurie de médicaments : Trouver d'urgence le bon remède, rapport de commission d'enquête, n° 828 (2022-2023), tome I, déposé le 4 juillet 2023.
* 1067 Pénurie de médicaments : Trouver d'urgence le bon remède, rapport de commission d'enquête, n° 828 (2022-2023), tome I, déposé le 4 juillet 2023.
* 1068 Cette mesure avait été introduite par un amendement du rapporteur général, M. Olivier Véran, en première lecture à l'Assemblée nationale.
* 1069 Article 57 de la loi n° 2022-1616 du 23 décembre 2022.
* 1070 Article 1 de l'arrêté du 16 octobre 2020 fixant les spécifications des médicaments à base de cannabis utilisés pendant l'expérimentation prévue à l'article 43 de la loi n° 2019-1446 du 24 décembre 2019 de financement de la sécurité sociale pour 2020, les conditions de leur mise à disposition ainsi que les indications thérapeutiques ou situations cliniques dans lesquelles ils seront utilisés
* 1071 « Suivi de l'expérimentation française de l'usage médical du cannabis », compte-rendu du 15 décembre 2022, Comité scientifique temporaire, ANSM.
* 1072 Article 5 bis de l'arrêté du 29 décembre 2020 fixant les modalités de participation des médecins et pharmaciens volontaires intervenant dans l'expérimentation prévue à l'article 43 de la loi n° 2019-1446 de financement de la sécurité sociale pour 2020 ainsi que les conditions de formation préalable obligatoire et de rémunération des professionnels de santé participant à cette expérimentation.
* 1073 Cette article liste et définit certains médicaments, parmi lesquels par exemple les préparations magistrales (1°), les préparations hospitalières (2°), les préparations officinales (3°), les médicaments de thérapie innovante (17°), les médicaments dérivés du sang (18°).
* 1074 Article L. 5121-5 du code de la santé publique.
* 1075 Sur ce point, voir l'article L. 5121-15 du même code qui se trouve rétabli.
* 1076 Article L. 5121-8 du code de la santé publique.
* 1077 Article L. 5121-9-1 du code de la santé publique.
* 1078 Article L. 5121-12 du code de la santé publique.
* 1079 Article L. 5121-12-1 du code de la santé publique.
* 1080 Exposé des motifs de l'amendement n° 3298 déposé par le Gouvernement à l'Assemblée nationale.
* 1081 Nocivité du médicament ; évaluation défavorable du rapport bénéficies-risques ; composition qualitative et quantitative non conforme ; non-respect de certaines de ses obligations par le titulaire, notamment en matière de pharmacovigilance.
* 1082 Ces dispositions sont prévues en miroir de l'article L. 5121-14-2 du code de la santé publique, qui confère la même compétence à l'ANSM pour les spécialités pharmaceutiques.
* 1083 Information aux utilisateurs mise en ligne le 31 mars 2023 sur le site internet de l'ANSM.