







CHAPITRE III
RÉNOVER LA RÉGULATION DES
DÉPENSES
DE PRODUITS DE SANTÉ
Article 33
Innovation numérique et médicaments
Cet article prévoit plusieurs mesures destinées à renforcer l'accès précoce des patients à l'innovation :
- il apporte plusieurs compléments et clarifications aux dispositifs d'accès précoce et d'accès compassionnel afin d'en renforcer l'opérationnalité et de garantir la continuité des traitements initiés ;
- il améliore les conditions de prise en charge des médicaments de thérapie innovante ;
- il crée un dispositif de prise en charge anticipée des solutions numériques innovantes en santé ;
- il renforce la prévisibilité et la lisibilité du forfait innovation pour la prise en charge dérogatoire des dispositifs médicaux et actes innovants ;
- il simplifie les modalités de fixation des prix des médicaments rétrocédables ;
- il supprime le coefficient de minoration des spécialités pharmaceutiques en soins de suite et de réadaptation.
La commission vous demande d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - Le dispositif proposé
A. Plusieurs mesures disparates concourant au meilleur accès des patients aux innovations
L'article 33 du PLFSS pour 2022 apporte plusieurs compléments et clarifications aux dispositifs d'accès précoce et d'accès compassionnel, issus de l'article 78 de la loi de financement de la sécurité sociale pour 2021 375 ( * ) , afin d'en renforcer l'opérationnalité et l'efficacité.
1. Le maintien de l'accès compassionnel à un médicament en dépit de la mise en place d'une recherche impliquant la personne humaine
En l'état du droit en vigueur, trois conditions cumulatives restreignent l'accès compassionnel, qui peut prendre la forme soit d'une autorisation d'accès compassionnel (AAC) à la demande d'un médecin prescripteur sur autorisation de l'agence nationale de sécurité du médicament et des produits de santé (ANSM), soit d'un cadre de prescription compassionnel (CPC) établi par l'ANSM de sa propre initiative ou à la demande des ministres chargés de la santé ou de la sécurité sociale :
- le médicament ne doit pas faire l'objet d'une recherche impliquant la personne humaine (RIPH) à des fins commerciales ;
- il n'existe pas de traitement approprié ;
- l'efficacité et la sécurité du médicament sont présumées au regard des données cliniques disponibles.
L'article L. 5121-12-1 du code de la santé publique prévoit néanmoins, à l'heure actuelle, qu'un médicament faisant l'objet à un stade très précoce d'une RIPH dans l'indication considérée peut bénéficier d'une AAC sous réserve que l'entreprise exploitant le médicament dépose, dans un délai qui a été fixé par décret 376 ( * ) à 12 mois à compter de l'octroi de l'AAC et à 18 mois lorsque l'indication porte sur une maladie rare, une demande d'accès précoce au titre de l'article L. 5121-12 dans l'indication considérée.
Le 1° du I de l'article 33 du PLFSS pour 2022 modifie l'article L. 5121-12-1 précité afin de permettre le maintien de l'accès compassionnel à un médicament dans le souci d'assurer la continuité des traitements, même lorsqu'une RIPH à promotion industrielle a été engagée, le temps qu'une demande de droit commun intervienne :
- une AAC pourra ainsi être maintenue ou renouvelée par l'ANSM pour tenir compte de la situation particulière d'un patient et pour une durée maximale prévue par décret, même si l'entreprise exploitant le médicament et ayant lancé une RIPH à des fins commerciales n'a pas déposé une demande d'autorisation précoce (AAP) à l'issue des délais réglementaires ;
- un CPC pourra être maintenu ou renouvelé pour des motifs de santé publique.
En outre, le 1° du I de l'article 33 du PLFSS pour 2022 clarifie, au sein de l'article L. 5121-12-1 du code de la santé publique, la terminologie applicables aux entreprises responsables en matière d'accès compassionnel, afin de distinguer celles qui sont titulaires de l'autorisation de mise sur le marché (AMM), responsables en cas de cadre de prescription compassionnelle pour des médicaments disposant déjà d'une AMM dans une autre indication, et celles qui sont titulaires des droits d'exploitation, responsables en cas d'autorisation d'accès compassionnel pour des médicaments ne disposant d'aucune AMM.
2. La clarification des modalités de prise en charge des phases de continuité de traitement au titre de l'accès précoce ou compassionnel
a) Le maintien de la prise en charge des phases de continuité de traitement au titre de l'accès précoce ou compassionnel dans le cadre de la « liste collectivités »
L'article L. 5123-2 du code de la santé publique conditionne l'achat, la fourniture, la prise en charge et l'utilisation de médicaments en milieu hospitalier à leur inscription sur la liste des spécialités agréées aux collectivités, dite « liste collectivités ». Il est néanmoins prévu, en l'état du droit en vigueur, que peuvent être pris en charge des médicaments ne figurant pas sur la « liste collectivités » dans deux hypothèses : lorsqu'ils ont fait l'objet d'autorisations ou de cadres de prescription compassionnelle ou lorsqu'ils bénéficient d'une autorisation d'importation en réponse à une rupture ou à un risque de rupture de stock ou à un arrêt de commercialisation.
Le a du 2° du I de l'article 33 du PLFSS pour 2022 complète les cas dans lesquels des médicaments peuvent être achetés, fournis, utilisés et pris en charge par les collectivités publiques sans figurer sur la « liste collectivités ». S'ajoutent ainsi aux médicaments faisant l'objet d'une AAP, d'une AAC ou d'un CPC et aux médicaments bénéficiant d'une autorisation d'importation pour faire face à une tension ou une rupture d'approvisionnement :
- les médicaments fournis aux établissements de santé par les laboratoires au titre de leur obligation d'assurer la continuité des traitements initiés dans le cadre des dispositifs d'accès précoce 377 ( * ) et d'accès compassionnel 378 ( * ) . Cette continuité doit en effet être assurée pendant toute la prise en charge dérogatoire par l'assurance maladie, puis, à l'issue de cette prise en charge, pour une durée minimale fixée par décret et ne pouvant excéder un an ;
- les médicaments faisant l'objet d'une autorisation de médicaments de thérapie innovante préparés ponctuellement (MTIPP) par l'ANSM ;
- les médicaments dont l'utilisation et la prise en charge, dans une indication pour laquelle ils ne disposent pas d'une AMM, sont autorisés par les autorités ministérielles lorsqu'ils sont administrés en association, dans l'indication considérée, avec un autre médicament disposant lui spécifiquement d'une AMM dans cette indication. Ces médicaments, dits à « AMM miroir » car bénéficiant indirectement de l'AMM du médicament auquel ils sont associés, font l'objet d'une régularisation des modalités de leur prise en charge précisées par le nouvel article L. 162-18-1 du code de la sécurité sociale créé par l'article 34 du PLFSS pour 2022.
Par ailleurs, le 4° du II de l'article 33 du PLFSS pour 2022 procède, au sein de l'article L. 162-16-5-4 relatif aux modalités de prise en charge des médicaments ayant fait l'objet d'une AAP au titre des continuités de traitement, à quelques clarifications rédactionnelles ( a et b ) et précise les règles encadrant les phases de continuité de traitement :
- outre les conditions de prise en charge 379 ( * ) , il apparaît nécessaire de définir les conditions de prescription et de dispensation des médicaments ayant bénéficié d'une AAP dans les phases de continuité de traitement : ainsi, le c précise que, si le médicament faisant l'objet d'une prise en charge au titre de l'AAP n'est inscrit ni sur la « liste ville » ni sur la « liste collectivités » pour l'indication considérée, ce sont les dernières conditions de prescription et de dispensation prévues dans le cadre de l'accès précoce qui s'appliqueront pendant la période de continuité de traitement ;
- le d transfère du CEPS aux ministres de la santé et de la sécurité sociale la possibilité de prononcer une pénalité financière à l'encontre d'un laboratoire qui méconnaîtrait ses obligations en matière de continuité des traitements initiés. Il est précisé que la pénalité sera recouvrée par les unions de recouvrement des cotisations de sécurité sociale et d'allocations familiales (Urssaf) et que son produit sera affecté à la CNAM.
b) Le maintien de la prise en charge des phases de continuité de traitement au titre de l'accès précoce ou compassionnel dans le cadre de la liste de rétrocession
L'article L. 5126-6 du code de la santé publique prévoit la possibilité pour certains établissements de santé et groupements de coopération sanitaire disposant d'une pharmacie à usage intérieur (PUI) de vendre au détail au public des médicaments réservés à l'usage hospitalier 380 ( * ) et qui présentent des contraintes particulières de distribution, de dispensation ou d'administration ou requièrent un suivi de la prescription ou de la délivrance.
Les médicaments concernés peuvent être rétrocédés par les PUI aux patients non hospitalisés, par dérogation ou en complément du circuit de droit commun des officines de ville, et peuvent notamment faire l'objet d'une délivrance à domicile, à la condition d'être inscrits sur une liste établie par l'ANSM, dite « liste de rétrocession ». La rétrocession est décidée pour des raisons de santé publique et concerne notamment des médicaments dérivés du sang, de médicaments pour la prise en charge des hépatites B ou C chroniques, de médicaments orphelins, de médicaments anticancéreux ou encore d'antirétroviraux, d'antibiotiques, d'antifongiques...
Depuis la réforme des dispositifs d'accès précoce et d'accès compassionnel en LFSS pour 2021, l'article L. 5126-6 précité prévoit, depuis le 1 er juillet 2021, que certains médicaments ne figurant pas sur la liste de rétrocession sont néanmoins réputés inscrits sur cette liste : il s'agit des médicaments non classés dans la catégorie des médicaments réservés à l'usage hospitalier et qui font l'objet d'une autorisation ou d'un cadre de prescription compassionnelle.
Le 3° du I de l'article 33 du PLFSS pour 2022 complète les cas dans lesquels des médicaments non réservés à l'usage hospitalier sont réputés inscrits sur la liste de rétrocession afin d'y inclure les médicaments fournis aux établissements de santé par les laboratoires au titre de leur obligation d'assurer la continuité des traitements initiés dans le cadre des dispositifs d'accès précoce et d'accès compassionnel.
3. La rationalisation de la gestion de la liste de rétrocession
a) La facilitation de l'inscription des médicaments rétrocédables sur la « liste collectivités »
Les médicaments rétrocédables étant délivrés à des patients ambulatoires, leur dispensation est imputée non pas sur le budget de l'établissement mais sur les dépenses d'assurance maladie dès lors qu'ils sont délivrés à des patients ambulatoires.
L'article 29 de la loi d'accélération et de simplification de l'action publique (ASAP) du 7 décembre 2020 381 ( * ) a transféré la gestion de la liste de rétrocession du ministre chargé de la santé au directeur général de l'ANSM. Ce transfert est entré en vigueur le 1 er octobre 2021, les nouvelles compétences de l'agence ne pouvant toutefois être complètement exercées à ce stade, dans l'attente de la publication du décret d'application en cours d'examen par le Conseil d'État.
En tout état de cause, ce transfert a pour conséquence de modifier les modalités d'inscription d'un médicament sur la liste de rétrocession. En effet, l'ANSM est d'ores et déjà appelée à fixer les conditions de prescription et de délivrance (CPD) d'un médicament dès que celui-ci a obtenu son AMM. L'inscription par l'ANSM du médicament sur la liste de rétrocession intervient ainsi logiquement dès l'octroi de l'AMM, à l'occasion de la définition des CPD par l'agence.
Or, en l'état du droit en vigueur, toute demande d'inscription d'un médicament non classé dans la catégorie des médicaments réservés à l'usage hospitalier sur la « liste collectivités » est conditionnée, par l'article L. 5123-2 du code de la santé publique, au dépôt par le laboratoire d'une demande d'inscription sur la liste des spécialités remboursables par l'assurance maladie et dispensées en ville, dite « liste ville » 382 ( * ) . Cette condition ne se justifie logiquement pas pour l'inscription des médicaments inscrits par l'ANSM sur la liste de rétrocession au titre de leur AMM : la procédure d'inscription sur la liste de rétrocession est en effet entièrement déconnectée de l'évaluation des médicaments par la HAS en vue de leur prise en charge au titre de la « liste collectivités » ou de la « liste ville ». En conséquence, le b du 2° du I de l'article 33 du PLFSS pour 2022 lève cette condition pour les médicaments classés comme rétrocédables par l'ANSM dès l'obtention de leur AMM.
b) La révision de la procédure de fixation du prix des médicaments inscrits sur la liste de rétrocession
Par ailleurs, dans la mesure où il revient désormais à l'ANSM de décider du circuit de dispensation entre officines de ville et PUI des médicaments rétrocédables, la procédure de prise en charge et de fixation du prix de ces médicaments doit être revue.
Pour mémoire, à l'heure actuelle, les conditions de prise en charge de ces médicaments sont précisées par un arrêté de prise en charge au titre de la rétrocession, un avis de prix de cession du CEPS et un taux de prise en charge arrêté par l'union nationale des caisses d'assurance maladie (Uncam). En application de l'article L. 162-16-5 du code de la sécurité sociale, le prix de cession au public d'un médicament rétrocédable est fixé par convention entre l'entreprise exploitant le médicament et le CEPS ou, à défaut, par décision de ce dernier, au plus tard dans un délai de 75 jours après son inscription sur la liste des médicaments rétrocédables 383 ( * ) . Ces dispositions n'ont pas été modifiées à l'occasion du transfert à l'ANSM de la gestion de la liste de rétrocession par la loi ASAP du 7 décembre 2020.
Or le délai de 75 jours entre l'inscription du médicament sur la liste de rétrocession et la fixation de son prix par le CEPS ne se justifie plus. En effet, cette inscription, désormais opérée par l'ANSM, intervient dès l'octroi de l'AMM et la fixation des conditions de prescription et de délivrance par l'agence, et non plus après la publication de l'avis de la commission de transparence de la HAS en vue de l'inscription du médicament sur la seule « liste collectivités ».
En conséquence, le 2° du II de l'article 33 du PLFSS pour 2022 modifie l'article L. 162-16-5 du code de la sécurité sociale afin de dissocier la négociation du prix de cession du médicament rétrocédable entre le CEPS et l'entreprise exploitant ou important le médicament de l'inscription de ce dernier sur la liste de rétrocession par l'ANSM. Il est précisé, dans l'étude d'impact annexée au PLFSS, que l'article R. 163-9 du code de la sécurité sociale sera modifié par la suite afin de prévoir, pour la fixation du prix de cession, un délai de 180 jours à compter du dépôt de la demande d'inscription du médicament sur la liste de rétrocession, dans le respect des règles européennes 384 ( * ) .
4. La création d'un cadre de prise en charge spécifique pour les médicaments de thérapie innovante préparés ponctuellement
Les médicaments de thérapie innovante (MTI) correspondent, en droit communautaire, à l'un des quatre types de médicaments suivants : les médicaments de thérapie génique, les médicaments de thérapie cellulaire somatique, les produits issus de l'ingénierie cellulaire - qui permettent de régénérer, réparer ou remplacer des tissus humains - et les médicaments combinés de thérapie innovante - qui incluent des dispositifs médicaux ou des dispositifs médicaux implantables actifs -.
Le 17° de l'article L. 5121-1 du code de la santé publique définit le médicament de thérapie innovante préparé ponctuellement (MTI-PP) comme tout médicament de thérapie innovante au sens du droit européen « fabriqué en France selon des normes de qualité spécifiques et utilisé dans un hôpital en France, sous la responsabilité d'un médecin, pour exécuter une prescription médicale déterminée pour un produit spécialement conçu à l'intention d'un malade déterminé. » Au titre de l'exemption hospitalière définie en droit européen, de nombreux MTI peuvent ainsi continuer à être utilisés en l'absence d'AMM « à condition que le produit soit utilisé dans un hôpital pour un patient déterminé et sous la responsabilité professionnelle d'un médecin. » 385 ( * )
Les MTI-PP font l'objet d'une autorisation délivrée par l'ANSM et les conditions d'autorisation de ces médicaments sont précisées aux articles R. 5121-209 et suivants du code de la santé publique. En application de l'article L. 4211-9-1 du même code, les MTI-PP ne peuvent être préparés que par les établissements ou organismes autorisés à cet effet par le directeur général de l'ANSM. Selon des données rendues disponibles par l'ANSM 386 ( * ) , les huit établissements autorisés en 2020 à préparer des MTI-PP sont uniquement des établissements de santé. La diminution du nombre d'établissements autorisés s'explique principalement par l'ouverture d'établissements pharmaceutiques par l'établissement français du sang (EFS) et le centre de transfusion sanguine des armées (CTSA), ainsi que par la publication en mai 2019 de bonnes pratiques de fabrication auxquelles les établissements ont dû se conformer.
Évolution des autorisations pour l'établissement de MTI-PP
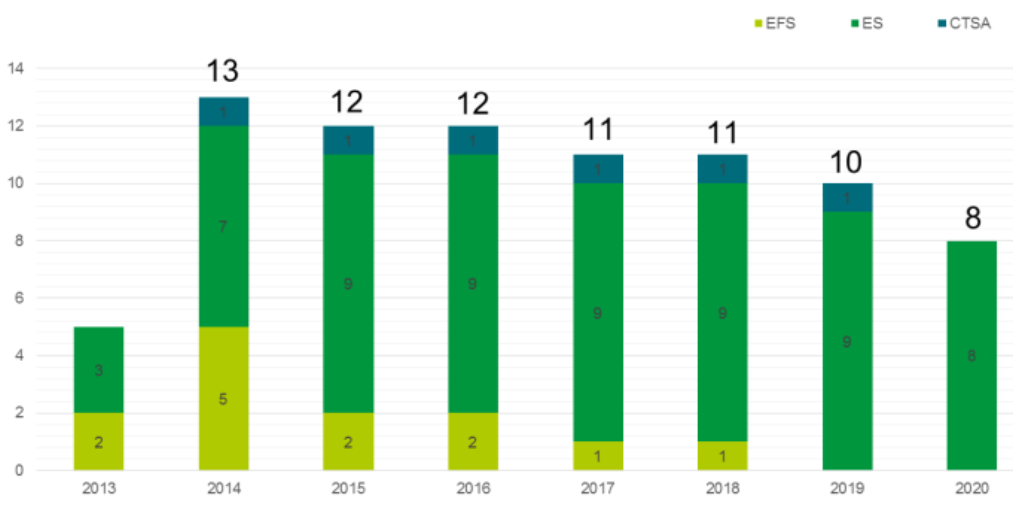
Source : Agence nationale de sécurité du médicament et des produits de santé
Selon l'étude d'impact annexée au PLFSS, 25 patients auraient été traités au cours de la dernière décennie à partir de quatre autorisations de MTI-PP. Des données issues du comité d'interface de l'ANSM sur les MTI évoquent, pour le seul centre hospitalier universitaire (CHU) de Nantes, la fabrication depuis 2014 de 76 lots de MTI-PP pour traiter 69 patients. Pour la seule année 2020, trois MTI ont été autorisés, dont deux avec une AMM conditionnelle, et deux demandes d'AMM ont été retirées par les demandeurs.
Nombre d'unités de MTI-PP distribuées et de patients traités
|
N° du MTI-PP (date d'autorisation) |
Nombre d'unités distribuées (chiffres extraits des rapports annuels d'activité des établissements) |
Nombre de patients traités depuis
l'autorisation
|
||
|
2019 |
2020 |
|||
|
HOSPICES CIVILS
|
MTI-PP 008 (23/05/2016)
Feuillet épidermique
|
250 |
482 |
27 |
|
EFS ÎLE DE FRANCE, Créteil |
MTI-PP 004 (06/02/2019) Moelle osseuse autologue concentrée à usage orthopédique |
11 |
40 |
65 |
|
CHU DE NANCY |
MTI-PP 009 (17/02/2016) Lymphocytes T anti-adénovirus allogéniques |
9 |
10 |
13 |
|
CTSA |
MTI-PP 007 (15/10/2015) Concentré de cellules stromales mésenchymateuses autologues amplifiées in vitro |
Absence de données |
5 |
8 |
Source : Agence nationale de sécurité du médicament et des produits de santé
À l'heure actuelle, en l'absence de prise en charge spécifique par l'assurance maladie, le coût de la production des MTI-PP est intégralement supporté par les établissements de santé. D'après l'étude d'impact annexée au PLFSS, « le coût de traitement associé aux MTI-PP sur une plateforme académique et mobilisant la technologie des CAR-T-cells représente un coût de l'ordre de 150 000 euros. »
Dès lors, le 5° du II de l'article 33 du PLFSS pour 2022 insère dans le code de la sécurité sociale un nouvel article L. 162-16-5-5 prévoyant la prise en charge des MTI-PP par l'assurance maladie : cette prise en charge s'effectuera sur une base forfaitaire annuelle par patient définie par arrêté des ministres de la santé et de la sécurité sociale suivant des modalités précisées par décret en Conseil d'État. Par coordination, le 6° du II de l'article 33 du PLFSS pour 2022 prévoit, à l'article L. 162-22-7-3 du code de la sécurité sociale, que cette prise en charge des MTI-PP par l'assurance maladie, de même que la prise en charge des médicaments disposant d'une « AMM miroir » s'effectueront en sus des prestations d'hospitalisation.
Partant de l'hypothèse de cinq patients traités par MTI-PP en 2022 et de six patients supplémentaires par an les années suivantes, l'étude d'impact estime le coût de la prise en charge des MTI-PP pour l'assurance maladie à trois millions d'euros en 2022, à quatre millions d'euros en 2023, à 12 millions d'euros en 2024 et à 17 millions d'euros en 2025.
5. La suppression du coefficient de minoration des spécialités pharmaceutiques en soins de suite et de réadaptation
Les soins de suite et de réadaptation (SSR) sont financés selon des modalités fixées par les articles L. 162-23 et suivants du code de la sécurité sociale. Un objectif de dépenses d'assurance maladie afférent aux activités de SSR est ainsi arrêté chaque année par l'État en fonction de l'Ondam. Cet objectif distingue les parts afférentes :
- aux dépenses relatives au financement de la liste des spécialités pharmaceutiques qui, en raison de leur coût, peuvent être prises en charge en sus des prestations d'hospitalisation (article L. 162-23-6 du code de la sécurité sociale) ;
- à la dotation nationale affectée au financement des missions d'intérêt général et d'aide à la contractualisation des établissements participant aux activités de SSR (article L. 162-23-8 du code de la sécurité sociale).
En application de l'article L. 162-23-6 du code de la sécurité sociale, les remboursements perçus par les établissements au titre de la liste des médicaments onéreux peuvent être minorés par l'application d'un coefficient censé concourir au respect de l'objectif de dépenses en matière de SSR affecté à ce poste de dépenses.
Toutefois, ce mécanisme prudentiel semble peu justifié à deux titres :
- d'une part, il n'a jamais été appliqué à ce jour : en application du V de l'article 78 de la loi de financement de la sécurité sociale pour 2016 et par dérogation à l'article L. 162-23-6 du code de la sécurité sociale, les dépenses relatives à la consommation des molécules onéreuses font l'objet, jusqu'au 31 décembre 2021, d'un montant dédié au sein de la dotation annuelle 387 ( * ) de financement allouée aux établissements au titre de leurs activités de SSR ;
- d'autre part, comme le rappelle l'étude d'impact annexée au PLFSS, l'application d'un coefficient de minoration à la prise en charge des molécules onéreuses et donc la perspective d'un remboursement non intégral pourrait exercer un effet désincitatif au transfert des patients du champ « Médecine, chirurgie, obstétrique » (MCO) vers le champ SSR. En outre, l'existence de ce coefficient place le champ SSR dans une situation d'iniquité vis-à-vis du champ MCO pour lequel un tel coefficient n'existe pas.
Pour l'ensemble de ces raisons, le 8° du II de l'article 33 du PLFSS pour 2022 supprime le coefficient de minoration du remboursement des spécialités pharmaceutiques en SSR à l'article L. 162-23-6 du code de la sécurité sociale. En conséquence, le 7° du II de l'article 33 du PLFSS pour 2022 procède à une coordination à l'article L. 162-23-4 du même code.
6. Le renforcement de la lisibilité et de la prévisibilité du forfait innovation
Créé par la loi de financement de la sécurité sociale pour 2009 388 ( * ) , le forfait innovation permet, en application de l'article L. 165-1-1 du code de la sécurité sociale, la prise en charge dérogatoire des dispositifs médicaux et d'actes innovants. Il peut être mobilisé tant par les industriels que par les sociétés savantes. S'il contribue à faciliter l'accès précoce du patient à une technologie de santé innovante, il vise également à développer les données cliniques ou médico-économiques encore manquantes sur le traitement innovant en subordonnant sa mise en oeuvre à la conduite d'études. Les résultats de ces études doivent permettre d'éclairer les décisions futures en matière de prise en charge pérenne par la collectivité.
Procédure de demande de prise en charge au titre du forfait innovation

Source : Ministère des solidarités et de la santé
( http://solidarites-sante.gouv.fr/IMG/pdf/forfait_innovation_procedure_prise_en_charge.pdf )
En juin 2020, la HAS a ouvert le portail électronique « Sésame » afin de simplifier les modalités de dépôt des demandes et a appelé les industriels et les sociétés savantes « à recourir davantage au forfait innovation ». Selon un bilan de la HAS, sur la période 2015-2020, sur 29 dossiers déposés, 12 seulement ont été déclarés éligibles.
Bilan des dossiers de forfait innovation traités par la Haute Autorité de santé
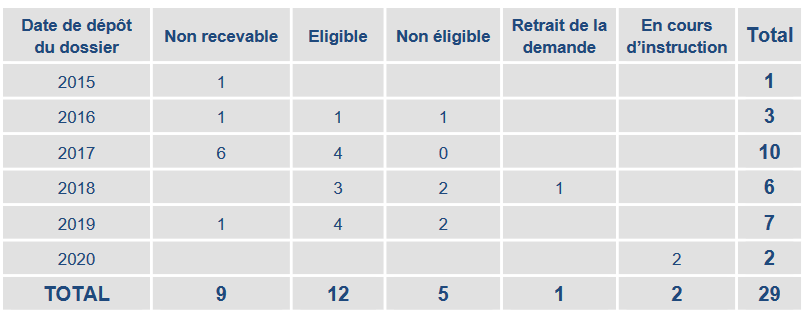
Source : Haute Autorité de santé, communiqué de presse du 30 juin 2020
Bilan du forfait innovation
Selon des informations transmises par la direction de la sécurité sociale, à ce jour, le forfait innovation a permis de financer 14 projets pour un total de 69 millions d'euros. Ces financements permettent d'accompagner 13 000 patients dans le cadre d'essais cliniques dont les critères ont été évalués et validés par la HAS afin de démontrer l'intérêt de dispositifs médicaux ou de diagnostic.
Ces projets innovants concernent aussi bien des dispositifs de diagnostic du SARS-CoV-2, ou de test fonctionnel dans le cancer colorectal métastatique non opérable d'emblée destiné à personnaliser les protocoles de chimiothérapies associées ou non à une thérapie ciblée, que des dispositifs médicaux à visée thérapeutique, tel que le traitement de la douleur des patients souffrant de crises d'algie vasculaire de la face insuffisamment soulagés par les moyens thérapeutiques existants ou le traitement de l'insuffisance cardiaque par un coeur artificiel.
Un des dispositifs médicaux ayant bénéficié du financement par le forfait innovation a été évalué par la HAS en vue d'une prise en charge dans le droit commun au titre de l'article L. 165-1 du code de la sécurité sociale. La HAS lui a octroyé un service médical rendu suffisant et une amélioration du service médical rendu de niveau III ou modérée par rapport à l'absence d'alternative. Ce dispositif, ARGUS II, est destiné compenser le handicap lié à la perte de vision induite par la dégénérescence rétinienne en stade avancée.
Source : Direction de la sécurité sociale
Afin de renforcer la visibilité des demandeurs sur le niveau de compensation financière qui pourrait leur être accordée dans le cadre du forfait innovation, le 9° de l'article 33 du PLFSS pour 2022 précise, à l'article L. 165-1-1 du code de la sécurité sociale, une série de critères susceptibles de déterminer le niveau de prise en charge du produit de santé, de la prestation ou de l'acte innovant.
Sans que la liste envisagée soit exhaustive - l'adverbe « notamment » précédant l'énumération des critères -, il est prévu de tenir compte :
- des tarifs des produits et prestations à visée thérapeutique comparable ;
- des tarifs constatés dans d'autres pays européens ;
- des volumes de ventes prévus des produits ou prestations et des montants remboursés par l'assurance maladie prévus ;
- des actes existants déjà pris en charge.
En outre, l'article L. 165-1-1 du code de la sécurité sociale est complété par des dispositions visant à renforcer les engagements de l'exploitant d'un produit en contrepartie de la compensation versée par l'assurance maladie dans le cadre du forfait innovation. Il devra ainsi mener à terme l'étude clinique ou médico-économique, sauf en cas de risque avéré pour la sécurité des patients ou d'interruption anticipée de la prise en charge justifiée par des données démontrant l'existence ou l'absence de bénéfice clinique ou médico-économique. L'exploitant sera également tenu de déposer une demande d'inscription sur la liste des produits et prestations (LPP) 389 ( * ) dans un délai d'un an après la fin de l'étude si les résultats de cette dernière sont positifs.
En cas de manquement à ses obligations, l'exploitant pourra se voir appliquer une pénalité financière ne pouvant dépasser 30 % du montant hors taxe perçu dans le cadre du forfait innovation au titre du dispositif médical.
B. La création d'un dispositif de prise en charge anticipée des innovations dans le champ du numérique en santé
À la suite du 9 e conseil stratégique des industries de santé (CSIS), le Gouvernement s'est engagé, dans le cadre du plan « Innovation santé 2030 », à lancer une réflexion sur « la mise en place d'un accès dérogatoire aux dispositifs médicaux numériques, afin de faciliter leur accès au marché. » Le 1° du II de l'article 33 du PLFSS pour 2022 institue ainsi, au sein d'un nouvel article L. 162-1-23 du code de la sécurité sociale, un système de prise en charge anticipée des dispositifs médicaux numériques ou d'activités de télésurveillance médicale.
1. Une prise en charge anticipée d'un an non renouvelable limitée aux dispositifs médicaux numériques et aux activités de télésurveillance médicale
En application du I du nouvel article L. 162-1-23 du code de la sécurité sociale, cette prise en charge dérogatoire par l'assurance maladie, d'une durée limitée à un an non renouvelable, concernera, dans une indication particulière :
- soit un dispositif médical numérique 390 ( * ) à visée thérapeutique. Il s'agit des thérapies numériques, également connues sous le vocable anglo-saxon de « digital theapeutics » (DTx), notamment utilisées dans la prise en charge de certaines pathologies psychiatriques ;
- soit des activités de télésurveillance médicales, qui combinent la surveillance médicale et l'utilisation de dispositifs médicaux numériques, conformément régime de prise en charge de droit commun de ces activités institué par l'article 24 du PLFSS pour 2022.
Cet accès au remboursement anticipé est transitoire, les dispositifs ou activités concernés ayant vocation à être inscrits sur la LPP 391 ( * ) ou sur la liste des activités de télésurveillance instituée par l'article 24 du PLFSS pour 2022. Les obligations de déclaration préalable des opérateurs de télésurveillance médicale auprès de l'ARS compétente, prévues par l'article 24 du PLFSS pour 2022, seront également applicables aux activités de télésurveillance faisant l'objet d'une prise en charge anticipée.
2. Les critères de la prise en charge anticipée
En application du II du nouvel article L. 162-1-23 du code de la sécurité sociale, la prise en charge anticipée, sollicitée par l'exploitant du dispositif médical numérique - qu'il ait une visée thérapeutique ou qu'il soit utilisé dans le cadre d'une activité de télésurveillance -, sera décidée par les ministres de la santé et de la sécurité sociale après avis de la CNEDiMTS de la HAS. Elle est conditionnée à quatre critères cumulatifs :
- la caractère présumé innovant ;
- le marquage CE du dispositif médical numérique ;
- la conformité aux règles de protection des données personnelles et aux référentiels de sécurité et d'interopérabilité ;
- la capacité à exporter et échanger les données traitées dans des formats interopérables.
3. Un remboursement sur une base forfaitaire, subordonné à des engagements de la part du bénéficiaire
En application du III du nouvel article L. 162-1-23 du code de la sécurité sociale, la compensation versée au distributeur au détail du dispositif ou à l'opérateur de télésurveillance médicale prendra la forme d'un forfait fixé par arrêté ministériel, dans des conditions précisées par voie réglementaire, et ne pourra être cumulée avec d'autres modes de prise en charge prévus par le code de la sécurité sociale.
Le IV du nouvel article L. 162-1-23 du code de la sécurité sociale prévoit qu'en contrepartie de cette compensation, le bénéficiaire doit s'engager à :
- déposer une demande d'inscription sur la LPP ou la liste des activités de télésurveillance médicale dans des délais respectifs de six mois et neuf mois à compter de la prise en charge transitoire ;
- assurer la continuité des traitements ou de la surveillance médicale initiés, non seulement pendant la durée de la prise en charge transitoire mais également pendant une durée d'au moins six mois à compter de l'arrêt de cette prise en charge. Cette durée est néanmoins ramenée à 45 jours en cas de refus de l'inscription sur la LPP ou la liste des activités de télésurveillance médicale. Pendant la durée de continuité des traitements postérieure à la prise en charge anticipée, le remboursement est accordé dans les mêmes conditions que dans le cadre de la LPP ou de la liste des activités de télésurveillance ou, à défaut, dans les mêmes conditions que pendant la prise en charge anticipée.
Le V du nouvel article L. 162-1-23 du code de la sécurité sociale définit les cas dans lesquels la prise en charge anticipée est interrompue, qui sont au nombre de trois :
- lorsque l'engagement de déposer une demande d'inscription sur la LPP ou la liste des activités de télésurveillance médicale dans les délais légaux n'est pas respecté ;
- lorsque la décision d'inscription ou de refus d'inscription sur les listes de droit commun est prise et que le tarif de responsabilité est publié ;
- lorsque les critères de la prise en charge anticipée - hors, en toute logique, le critère de la présomption d'innovation - ne sont plus remplis.
Le VI du nouvel article L. 162-1-23 du code de la sécurité sociale définit les modalités de mise en oeuvre de la pénalité financière applicable à l'exploitant du dispositif médical numérique qui méconnaîtrait ses obligations de continuité des traitements. Cette pénalité ne peut dépasser 30 % du chiffre d'affaires hors taxe réalisé en France au titre du dispositif au cours des 24 mois précédant la constatation du manquement.
Enfin, le VII du nouvel article L. 162-1-23 du code de la sécurité sociale renvoie à un décret en Conseil d'État la définition des modalités d'application de ses dispositions.
II - Les modifications adoptées par l'Assemblée nationale
Outre plusieurs amendements rédactionnels, l'Assemblée nationale a adopté, à l'article 33 du PLFSS pour 2022, une série d'amendements ayant pour objectifs :
- de corriger, à l'initiative du rapporteur général et avec l'accord du Gouvernement, des erreurs dans la terminologie utilisée pour qualifier l'entreprise dont le médicament fait l'objet d'une autorisation ou d'un cadre de prescription au titre de l'accès compassionnel. Tant en matière de recueil des données concernant l'efficacité, les effets indésirables et les conditions réelles d'utilisation du médicament, qu'en matière de conditions de prise en charge dérogatoire, il convient en effet de distinguer le titulaire des droits d'exploitation du médicament, responsable en cas d'autorisation d'accès compassionnel pour un médicament ne disposant d'une AMM dans aucune indication, de l'entreprise qui exploite le médicament, responsable en cas de cadre de prescription compassionnelle qui porte sur un médicament disposant déjà d'une AMM dans une autre indication ;
- de garantir l'interopérabilité sémantique des dispositifs médicaux numériques. Un amendement de Mme Agnès Firmin Le Bodo, du groupe Agir ensemble, adopté avec les avis favorables de la commission et du Gouvernement, a ainsi complété les exigences d'interopérabilité applicables aux dispositifs médicaux numériques éligibles à une prise en charge anticipée, en précisant qu'ils devront permettre de télécharger des données structurées. Au-delà de l'interopérabilité technique, il convient en effet de d'assurer l'importation et l'exportation de données dans des formats garantissant leur bonne compréhension et leur exploitation par le patient et l'ensemble des acteurs de la prise en charge ;
- de prévoir, à l'initiative du rapporteur général et avec l'accord du Gouvernement, le non-cumul entre la prise en charge anticipée des dispositifs médicaux numériques et des activités de télésurveillance médicale et la prise en charge au titre des prestations d'hospitalisation donnant lieu à facturation, c'est-à-dire hors séjours tarifés en groupe homogène de séjour (GHS) ou en groupe homogène de tarifs (GHT) ;
- de conditionner la prise en charge dérogatoire à la vérification de l'utilisation effective du dispositif médical numérique par le patient et à la collecte de données en vie réelle. Un amendement de M. Jean-Louis Touraine, du groupe La République en marche, accepté par la commission et le Gouvernement, transpose ainsi au système de remboursement anticipé des dispositifs médicaux numériques et des activités de télésurveillance les mêmes exigences de contrôle de l'utilisation effective du dispositif et d'atteinte de résultats d'utilisation en vie réelle que celles prévues pour le régime de prise en charge de droit commun des activités de télésurveillance institué par l'article 24 du PLFSS pour 2022 ;
- de plafonner le prix d'achat d'une spécialité ayant fait l'objet d'une autorisation d'accès précoce pendant la période de continuité des traitements pour laquelle elle ne bénéficie plus de la prise en charge au titre de l'accès précoce. Un amendement du rapporteur général, ayant reçu l'avis favorable du Gouvernement, prévoit ainsi que l'exploitant doit permettre, en phase de continuité des traitements, l'achat de la spécialité à un prix qui n'excède pas son prix de référence pour la prise en charge au titre de l'accès précoce, le cas échéant au moyen de remises.
III - La position de la commission
Les thérapies numériques ou digitales (« Digital Therapeutics » - DTx) sont en passe de connaître un essor spectaculaire dans un contexte d'accroissement des pathologies chroniques. À titre d'exemple, l'application BlueStar® a fait l'objet d'une autorisation pour remboursement en 2013 par l'agence américaine de sécurité du médicament 392 ( * ) pour le suivi de patients atteints de diabète de type 2. Selon une étude de 2019 393 ( * ) produite par le cabinet de conseil Juniper Research , les thérapies numériques devraient représenter un marché de près de 32 milliards de dollars en 2024, contre 2,2 milliards de dollars en 2019. Les deux principales affections qui devraient tirer cette activité à la hausse seraient le diabète et l'obésité, qui concentreraient près de 19 milliards d'euros dans le marché des thérapies numériques.
La mise en place d'un mécanisme de prise en charge anticipée pour ces thérapies participe d'un meilleur accès des patients à l'innovation. Dans le domaine des thérapies digitales, l'exigence d'interopérabilité des données collectées et exportées par les dispositifs médicaux numériques est déterminante pour la qualité des soins dans le cadre d'une activité de télésurveillance : c'est à la condition d'être pleinement exploitables par l'équipe soignante et compréhensibles par le patient que les données produites par les dispositifs médicaux numériques apporteront une véritable valeur ajoutée au parcours de soins et renforceront l'autonomie du patient.
Partageant ce souci, l'Assemblée nationale a tenu à préciser, en première lecture, que les dispositifs médicaux numériques de télésurveillance doivent permettre de « télécharger des données structurées », afin de s'assurer que les données collectées ne soient pas simplement exportables dans un format PDF 394 ( * ) mais puissent être facilement exploitables par les équipes soignantes dans le cadre de la prise en charge. Toutefois, la rapporteure s'interroge sur la valeur ajoutée de cette précision, dès lors que l'article 33 du PLFSS pour 2022 prévoyait déjà, dans sa version initiale, l'exportation dans des formats interopérables et des interfaces pour l'échange de données avec des accessoires connectés.
En outre, selon des éléments transmis par la délégation du numérique en santé, l'expression de « données structurées » peut faire référence, dans le domaine de la santé, à des formats d'interopérabilité avec des représentations complexes, comme le montre le cadre d'interopérabilité des systèmes d'information de santé (CI-SIS). Or, potentiellement, des exports simples, de type « date et taux de glycémie », peuvent être utiles.
L'intention de la rédaction initiale, insistant sur la nécessité de « formats interopérables appropriés », consistait à obliger l'export interopérable de données soit suivant des standards « normés » (ou référentiels) - comme ceux du CI-SIS - qui peuvent être rendus opposables par arrêté 395 ( * ) , soit par des formats potentiellement simples et pragmatiques. Il convient de maintenir ces deux approches de l'export interopérable, si bien que la commission a adopté un amendement n° 171 substituant à l'obligation du téléchargement de données structurées celle de l'accès direct aux données.
Par coordination avec le régime de droit commun de prise en charge des dispositifs médicaux numériques de télésurveillance institué par l'article 24 du PLFSS pour 2022, la commission a adopté un amendement n° 172 précisant qu'en cas de refus du patient à la transmission par le professionnel de santé des données nécessaires à la mise en oeuvre du contrôle de cette utilisation effective, l'activité de télésurveillance ne pourra faire l'objet d'une prise en charge anticipée. Si cette prise en charge dérogatoire a déjà été enclenchée, elle sera alors suspendue. En outre, il est rappelé que le niveau de prise en charge pourra être modulé, voire suspendu en cas d'inutilisation répétée du dispositif.
Enfin, la commission s'interroge sur les raisons qui justifient de calculer le montant de la pénalité, en cas de manquement pour non-respect des engagements de l'industriel en matière de continuité des traitements, sur le chiffre d'affaires hors taxes (CAHT) réalisé au cours des 24 derniers mois précédant la constatation du manquement. Ces dispositions sont vraisemblablement inspirées du calcul des pénalités applicables 396 ( * ) aux industriels exploitant un médicament bénéficiant d'une autorisation d'accès précoce.
Toutefois, il convient de rappeler que les autorisations d'accès précoce ne sont pas limitées dans le temps et prennent fin lorsque l'indication est inscrite, au titre de l'AMM, sur une liste de médicaments remboursables. Or la prise en charge anticipée d'un dispositif médical numérique de télésurveillance innovant est limitée à un an, non renouvelable.
En conséquence, la commission a adopté un amendement n° 173 prévoyant que le montant de la pénalité sera calculé sur le CAHT réalisé au cours des 18 derniers mois précédant la constatation du manquement : cette durée permet de tenir compte du fait, qu'au-delà des 12 mois de la prise en charge anticipée, l'entreprise s'engage à mettre à disposition le dispositif médical numérique pour une durée complémentaire de six mois au titre des continuités de traitement.
La commission a également adopté un amendement rédactionnel n° 170.
La commission vous demande d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article 34
Remises
unilatérales médicaments et évolutions relatives à
l'inscription
de certains dispositifs médicaux
Cet article prévoit une prise en charge de médicaments de la liste en sus pour des utilisations hors référentiel et adapte les règles applicables aux dispositifs médicaux en matière d'évaluation requise et d'inscription pour une indication donnée.
La commission vous demande d'adopter cet article sans modification.
I - Le dispositif proposé
A. Une régularisation de la prise en charge des médicaments utilisés en « AMM miroir » sur la liste en sus
1. Un usage fréquent de médicaments hors-référentiel au coût important pour l'assurance maladie
Comme l'indique l'évaluation préalable, la dépense remboursée associée à la prise en charge dans le cadre de la liste en sus était de 3,9 milliards d'euros pour les spécialités pharmaceutiques. Cette dépense connaît par ailleurs un taux de croissance annuel dynamique, de 6 % sur la période 2014-2019.
Or, il apparaît selon le Gouvernement que dans 20 % des cas, les indications sont associées à des utilisations non présentes dans le référentiel, la prescription étant faite en dehors de l'autorisation de mise sur le marché . Ces utilisations représenteraient une dépense pour l'assurance maladie estimée entre 600 et 700 millions d'euros pour des utilisations qui, légalement, ne sont pas pris en charge.
Le Gouvernement identifie particulièrement une pratique, qualifiée d' « AMM miroir », le laboratoire du médicament utilisé en association n'ayant souvent pas d'intérêt à déposer de nouvelle demande d'AMM.
L'AMM miroir correspond à une indication d'AMM attribuée pour un médicament A en association à un médicament B alors que le médicament B ne dispose pas d'AMM en association au médicament A.
Comme le précise l'évaluation préalable, « l'association des deux médicaments est en pratique prise en charge, alors qu'un des deux médicaments est hors référentiel et que son prix n'a pas été négocié au regard de cette nouvelle utilisation , puisque ce dernier ne bénéficie pas de l'AMM et que l'exploitant ne fait donc aucune demande d'inscription, tandis que des ventes seront pourtant réalisées dans cette indication » . Cependant, cette pratique financière est, comme le souligne le Gouvernement, sans impact sanitaire, l'usage répondant aux autorisations délivrées. La direction de la sécurité sociale estime à une dizaine le nombre de spécialités concernées, considérant ces types d'AMM fréquents.
Le présent article entend régulariser ces utilisations et prévoir, avec leur reconnaissance, un dispositif de remises ad hoc. L'impact financier de la mesure est estimé à 100 millions d'euros par an.
2. Une régularisation proposée en vue de valoriser ces utilisations dans les remises attendues
a) Une nouvelle inscription des indications hors référentiel
Le 3° modifie le code de la sécurité sociale , y insérant un nouvel article L. 162-18-1 afin de prévoir le nouveau dispositif de remises.
À son I , le nouvel article prévoit les caractéristiques de ce régime avec :
- la condition d'inscription sur liste en sus des prises en charges hospitalières ( 1° ) ;
- la possibilité pour cette spécialité d'être utilisée en association avec d'autres spécialités qui bénéficient pour ces indications, en association avec la spécialité, d'une autorisation de mise sur le marché et d'une inscription sur une liste de remboursements de droit commun ou d'une autorisation d'accès précoce ( 2° ) ;
- le fait de ne pas bénéficier, pour cette indication en association, d'une autorisation de mise sur le marché ou d'une autorisation d'accès précoce ou compassionnel (3°).
Ces conditions cumulatives emportent une obligation de déclaration au comité économique des produits de santé (CEPS) par l'exploitant, l'importateur ou le distributeur de la spécialité du chiffre d'affaires réalisé durant l'année passée au titre de cette spécialité, ce avant le 15 février ( dernier alinéa du I )
b) Une mécanisme ad hoc de remises
Le A du II organise un cadre d'autorisation des utilisations en association définies au I et de prise en charge de celles-ci. Cette reconnaissance peut intervenir sur demande des entreprises ou à l'initiative même des ministres chargés de la santé et de la sécurité sociale.
La prise en charge ainsi permise est cependant conditionnée à la précision lors de la facturation, de ce cadre d'utilisation de la spécialité. Le dernier alinéa du A prévoit le recouvrement de l'indu en cas de non-respect.
Le B du II prévoit un mécanisme de remises pour les médicaments utilisés en indication dans le cas où ils ne disposent pas, au titres des indications en association, d'une autorisation de mise sur le marché et d'une inscription sur une liste de remboursements ou d'une autorisation d'accès précoce.
Ces remises, versées annuellement, sont calculées sur la base du chiffre d'affaires hors taxes facturé aux établissements pour la spécialité au titre des indications en association.
Le deuxième alinéa du B du II prévoit que ces remises sont définies selon un barème de taux progressifs .
Enfin, les troisième et quatrième alinéas du même B précisent la déductibilité des remises conventionnelles ainsi que le calcul du chiffre d'affaires au regard de la part d'utilisation de cette spécialité réalisé dans les indications en association.
c) Conditionnement du remboursement
Le 1° assure les coordinations nécessaires concernant les règles de remboursement, en ajoutant la référence au nouvel article créé aux articles relatifs aux à la tarification et à la transmission de données conditionnant la prise en charge , qui prévoient également le recouvrement de l'indu en cas de non-respect. Il s'agit notamment d'assurer la transmission des données en vie réelle des indications utilisées hors référentiel.
Le 6° procède par ailleurs à une coordination relative au service du contrôle médical.
Le 2° corrige enfin une référence, réalisant une coordination lacunaire des dispositions de la LFSS pour 2021.
B. Une prise en charge des dispositifs médicaux sur indication pour la liste en sus
1. Une inscription des dispositifs médicaux sur la liste en sus sur indication
En cohérence avec la prise en charge applicable aux médicaments inscrits sur la liste en sus et alors que l'inscription sur cette liste répond à des évaluations données, le présent article entend préciser l'inscription et la facturation des dispositifs médicaux sur la liste en sus par indication .
Le 4° modifie à cette fin l'article L. 162-22-7 du code de la sécurité sociale .
2. Une simplification de l'inscription sur la liste « intra-GHS »
Certains dispositifs médicaux doivent, au regard de certaines caractéristiques, figurer sur une liste spécifique pour être utilisée par les établissements de santé.
Les dispositifs médicaux concernés par le dispositif « intra-GHS » appartiennent à des catégories homogènes de produits de santé déterminées par arrêtés des ministres chargés de la santé et de la sécurité sociale. Ces catégories de dispositifs médicaux sont déterminées au regard de leurs caractères invasifs ou des risques que ces derniers peuvent présenter pour la santé humaine.
Afin d'être inscrits sur la liste « intra-GHS », les dispositifs médicaux doivent faire l'objet au préalable d'une évaluation par la Commission nationale d'évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS) de la Haute autorité de santé (HAS).
Les dispositifs médicaux doivent justifier de l'une au moins des exigences suivantes : la validation de l'efficacité clinique des dispositifs, la définition de spécifications techniques particulières ou l'appréciation de leur efficience au regard des alternatives thérapeutiques disponibles.
Source : Ministère de la santé
Aux termes du III de l' article L. 165-11 du code de la sécurité sociale relatif à la prise en charge des dispositifs médicaux dans le cadre d'une utilisation par les établissements de santé, l'inscription sur la liste dite « intra-GHS » se fait après dépôt d'une demande auprès de la commission compétente de la Haute Autorité de santé, la CNEDiMTS .
Cependant, comme le souligne le Gouvernement, cette procédure est exigée alors même que des avis équivalents ont pu être rendus. Il s'agit donc de simplifier la procédure d'inscription sur la liste intra-GHS dans le cas d'une évaluation existante .
Ainsi, le 5° du présent article modifie le III précité afin de déroger à cette demande auprès de la commission de la HAS dans le cas d'une évaluation déjà rendue sur ces dispositifs au titre d'une demande d'inscription sur la liste en sus . Celle-ci doit être suffisamment récente et avoir démontré un service attendu ou rendu suffisant.
L'Assemblée nationale a adopté cet article ainsi modifié de corrections rédactionnelles.
III - La position de la commission
• La rapporteure soutient l'intention de cet article, qui vise, au bénéfice d'une reconnaissance effective des « AMM miroirs » , à ajuster la contribution des industriels au regard des utilisations faites des spécialités et des indications en association dont ils peuvent bénéficier. Elle émet une réserve sur la rédaction proposée et la bonne intégration de l'ensemble des dispositifs , notamment de la nouvelle prise en charge au titre de l'accès direct créé à l'article 36.
Elle rappelle en outre que cette reconnaissance et l'adaptation de cette prise en charge se font sans impact sur la sécurité sanitaire des patients .
Cependant, comme souligné par le Leem auprès de la rapporteure, certaines ventes en situation d' « AMM miroir » font l'objet de remises conventionnelles qui peuvent déjà fournir un outil de cadrage. Aussi, le Leem estime le dispositif de déduction des remises conventionnelles « ni approprié, ni pertinent » en cela qu'il ne la possible prise en compte déjà réalisée de l'« AMM miroir » au sein de ces dernières.
Si le Leem suggère un mécanisme d'exemption de remise obligatoire pour le chiffre d'affaires déjà visé par des remises conventionnelles, cette option n'apparaît à ce stade pas opportune à votre rapporteure : en effet, cela reviendrait à considérer la prise en compte déjà systématique des « AMM miroirs » dans l'ensemble des remises conventionnelles, ce qui ne semble pas correspondre à la réalité.
Concernant la prise en charge sur indication des dispositifs médicaux de la liste en sus , la rapporteure soutient cette disposition qui, comme le souligne le Snitem « donnera la possibilité d'un suivi plus fin à condition que cela ne débouche pas sur une obligation administrative supplémentaire pour les entreprises ».
La commission vous demande d'adopter cet article sans modification.
Article 34 bis
(nouveau)
Lutte contre les pénuries de dispositifs
médicaux
Cet article, inséré par l'Assemblée nationale, vise à prévoir un dispositif de lutte contre les pénuries dans le champ des dispositifs médicaux.
La commission vous demande d'adopter cet article modifié par l'amendement qu'elle a adopté tendant à clarifier la rédaction du dispositif et renforcer les moyens d'information et donc de supervision de l'ANSM.
I - Le dispositif proposé
Le présent article, adopté à l'initiative du Gouvernement, entend prévoir dans la loi un dispositif graduel d'anticipation et de gestion des pénuries en ce qui concerne les dispositifs médicaux (DM) et les dispositifs médicaux de diagnostic in vitro (DMDIV).
Les dispositions proposées reprennent l'esprit des dispositions applicables aux médicaments, aux ruptures d'approvisionnement de médicaments et aux médicaments d'intérêt thérapeutique majeur prévues aux articles L. 5121-29 et suivants du code de la santé publique.
A. Un dispositif d'anticipation et de gestion échelonnées
1. Une assise légale pour un dispositif en cours de mise en oeuvre
Dans l'exposé des motifs de son amendement, le Gouvernement indique que l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) a « mis en place une phase pilote, en lien avec les industriels, conduisant les industriels qui fabriquent et/ou mettent sur le marché des DM et des DMDIV considérés comme " indispensables " , et qui n'ont pas réussi à gérer eux-mêmes la situation, à signaler à l'ANSM toute rupture de stock ou d'approvisionnement afin que soient trouvées au plus vite des solutions alternatives pour les patients ».
Étape 1 : Gestion préventive par l'opérateur en lien avec les utilisateurs et les acheteurs
> Par tous les moyens à sa disposition, le fabricant met en oeuvre un plan d'action approprié pour éviter l'indisponibilité des produits : contingentement, augmentation des capacités de production, identification de solutions alternatives, y compris auprès d'entreprises concurrentes, etc . À ce stade, l'ANSM n'est pas sollicitée.
Étape 2 : Gestion par l'opérateur en lien avec l'ANSM
> Si les actions mises en oeuvre ne suffisent pas à rétablir la disponibilité des produits, le fabricant transmet à l'ANSM une déclaration de rupture. Nous viendrons en appui et nous participerons à l'identification de solutions, en lien avec notre réseau de partenaires, dans l'intérêt des patients. Dans un souci de gestion optimale et de transparence, nous publierons les informations utiles sur cette rupture.
Source : Agence nationale de sécurité du médicament et des produits de santé
Le Gouvernement entend ici « conforter cette mission » par le présent article de loi.
À cette fin, les 1° et 2° du présent article modifient le code de la santé publique afin d'y rétablir un article L . 5211-5-1 relatif aux dispositifs médicaux et insérer un article L. 5221-6-1 relatif aux dispositifs médicaux de diagnostic in vitro .
Les deux articles codifiés, identiques, prévoient un dispositif graduel d'information et de réaction face aux risques de rupture .
Leur I prévoit la qualification de certains dispositifs médicaux ou dispositifs médicaux in vitro comme « indispensables ». Cette qualification, établie sur la base de critères définis par décret , peut être faite par le fabricant, son mandataire, l'importateur ou le distributeur. À défaut, l'ANSM peut également y procéder.
Dans le cas d'un constat de risque de rupture, les fabricants, mandataires, importateurs ou distributeurs précédemment visés sont tenus, aux termes des II des deux articles, de « mettre en oeuvre toute mesure utile et nécessaire anticipée » dans le but d'éviter la rupture de disponibilité et d'assurer la continuité des soins dans l'intérêt des patients.
Leur III prescrit, dans le cas d'un échec des mesures évoquées à garantir la disponibilité, une déclaration auprès de l'ANSM du risque de rupture ou de la rupture .
Enfin, dans le cas où elle constate un risque de rupture ou une rupture face auxquels aucune mesure n'aurait été prise ou la déclaration obligatoire non satisfaite, leur IV donne mission à l'ANSM de prendre toute mesure utile aux mêmes fins de disponibilité des produits et de continuité des soins .
2. Un dispositif qui s'inscrit en parallèle d'une nouvelle régulation européenne
Enfin, si le Gouvernement souhaite ici combler une lacune en matière de dispositions propres aux dispositifs médicaux, il précise également rejoindre avec cet amendement le projet de règlement européen en cours de discussion et les missions prévues dans ce texte pour l'Agence européenne du médicament (EMA). Ce projet de règlement prévoit notamment l'identification et la surveillance de médicaments et dispositifs médicaux « critiques en cas d'urgence ».
Extraits de la proposition de règlement du parlement
européen
et du conseil relatif à un rôle renforcé
de l'Agence européenne des médicaments
dans la
préparation aux crises et la gestion de celles-ci en ce qui
concerne
les médicaments et les dispositifs médicaux
Considérant (12) Afin d'améliorer la préparation aux crises et la gestion de celles-ci en ce qui concerne les médicaments et les dispositifs médicaux et d'accroître la résilience et la solidarité dans toute l'Union, il y a lieu de préciser les procédures et les rôles et obligations respectifs des différentes entités concernées et impliquées . Le cadre devrait s'appuyer sur les solutions ad hoc qui ont déjà été trouvées dans le contexte de la riposte à la pandémie de covid-19.
(13) Un système harmonisé de surveillance des pénuries de médicaments et de dispositifs médicaux devrait être établi, ce qui facilitera un accès approprié aux médicaments et dispositifs médicaux critiques lors des urgences de santé publique et des événements majeurs susceptibles d'avoir une incidence grave sur la santé publique. Ce système devrait être complété par des structures améliorées afin de garantir une gestion appropriée des crises de santé publique, d'assurer une coordination et de fournir des avis concernant la recherche et le développement de médicaments susceptibles de répondre aux urgences de santé publique. Afin de faciliter la surveillance et la notification des pénuries réelles ou potentielles de médicaments et de dispositifs médicaux, l'Agence devrait pouvoir demander et obtenir des informations et des données auprès des titulaires d'autorisations de mise sur le marché, des fabricants et des États membres concernés par l'intermédiaire de points de contact désignés .
Article 24
Obligations pour les fabricants de dispositifs médicaux, les mandataires et les organismes notifiés
1. Afin de faciliter les activités de surveillance visées à l'article 21 et à la demande de l'Agence, les fabricants de dispositifs médicaux figurant sur la liste des dispositifs médicaux critiques en cas d'urgence de santé publique et, le cas échéant, les organismes notifiés concernés communiquent les informations demandées au plus tard à l'échéance fixée par l'Agence . Ils fournissent les informations demandées par l'intermédiaire des points de contact désignés conformément à l'article 23, paragraphe 2, et au moyen des méthodes et du système de notification établis en application de l'article 23, paragraphe 1. Chaque fois que cela est nécessaire, ils fournissent des mises à jour des informations. [...]
Article 25
Obligations pour les États membres en ce qui concerne la surveillance et l'atténuation des effets des pénuries de dispositifs médicaux
• 1. Afin de faciliter les activités de surveillance visées à l'article 21 et à la demande de l'Agence, les États membres, au plus tard à l'échéance fixée par l'Agence :
• a) fournissent l'ensemble d'informations demandé par l'Agence, y compris les informations relatives aux besoins de dispositifs médicaux figurant sur la liste des dispositifs médicaux critiques en cas d'urgence de santé publique, ainsi que les données disponibles et estimées concernant le volume de la demande , par l'intermédiaire de leur point de contact désigné et au moyen des méthodes et du système de notification établis au titre de l'article 23, paragraphe 1 ;
• b) mentionnent l'existence d'éventuelles informations confidentielles de nature commerciale et précisent les raisons d'une telle qualification ;
• c) mentionnent l'absence éventuelle d'informations demandées ainsi que tout retard de fourniture de ces informations par rapport au délai fixé par l'Agence. [...]
Sur ce projet, le Sénat a adopté une résolution européenne portant avis motivé 397 ( * ) considérant une contrariété au principe de subsidiarité .
B. Des manquements sanctionnés financièrement
Les 3° et 4° complètent respectivement les articles L. 5461-9 et L. 5462-8 relatifs aux manquements soumis à sanction financière , respectivement pour les dispositifs médicaux et les dispositifs médicaux de diagnostic in vitro .
Constituent ainsi des manquements le fait, pour les fabricants, de ne pas informer l'ANSM d'un risque de rupture ou de toute rupture dans la disponibilité des DM/DMDIV.
Enfin, complétant le III de l'article L. 5471-1 du code de la santé publique, le 5° prévoit l'échelle des sanctions financières créées , fixant un plafond de 150 000 euros pour une personne physique et 30 % du chiffre d'affaires , dans la limite d'un million d'euros, pour une personne morale.
II - La position de la commission
La crise sanitaire liée à l'épidémie de covid-19 a montré le caractère stratégique que pouvaient avoir tant certaines molécules qu'une série de dispositifs médicaux. Aussi, la rapporteure partage pleinement la préoccupation d'une meilleure gestion des risques de rupture en matière de dispositifs médicaux , sur le modèle des dispositifs existants pour les médicaments.
La rapporteure s'interroge cependant sur la rédaction retenue par le présent dispositif, notamment la notion de « non-disponibilité » non distinguée du risque de rupture ou de la rupture, et sur la gradation du mécanisme d'anticipation et d'intervention .
• Particulièrement, il apparaît que les exigences formulées à l'égard des fabricants, mandataires, importateurs ou distributeurs sont insuffisamment supervisées dès les premiers stades , à savoir l'identification même des dispositifs médicaux et les mesures préventives. Aussi, la commission a adopté un amendement n° 174 de la rapporteure visant à renforcer l'information de l'ANSM aux étapes d'anticipation des ruptures et à clarifier la rédaction de cet article.
Enfin, comme elle l'avait soulevé lors de l'examen de l'article 34 du projet de loi de financement pour 2020 relatif aux pénuries de médicaments, la commission s'interroge fortement sur la place d'un tel dispositif en PLFSS . Alors que la charge supplémentaire pour l'ANSM, estimée à 300 000 euros au titre des quatre ETP nécessaires, peut difficilement être retenue pour apprécier la recevabilité, la justification en PLFSS au titre des sanctions applicables est très discutable , les sanctions étant réputées, en termes de recevabilité financière, avoir un produit nul. La commission estime ainsi que le présent dispositif encourt un risque de censure par le Conseil constitutionnel .
La commission vous demande d'adopter cet article modifié par l'amendement qu'elle a adopté.
Article 35
Production en urgence de médicaments critiques et
règles relatives aux préparations magistrales et
hospitalières
Cet article propose de sécuriser la possibilité pour des pharmacies hospitalières et des établissements pharmaceutiques publics de produire des médicaments essentiels concernés par des difficultés d'approvisionnement ou pour faire face à une crise sanitaire grave. Il vise également à faciliter l'accès des patients à des préparations hospitalières ou magistrales, notamment en l'absence de traitement adapté ou disponible.
Favorable à ces mesures, la commission propose de modifier cet article par l'adoption d'un amendement tendant à préciser la possibilité, pour les pharmacies hospitalières et les établissements pharmaceutiques publics, de recourir au réseau d'officines sous-traitantes pour la production de médicaments critiques. En outre, elle a adopté un amendement visant à sécuriser le mode de financement de ces activités pour les établissements de santé.
I - Le dispositif proposé
A. La consécration de la possibilité pour des structures pharmaceutiques publiques de produire des médicaments critiques
Le phénomène des pénuries de médicaments, qui s'est amplifié au cours des dix dernières années, a été fortement aggravé par l'épidémie de covid-19. Sous l'effet des tensions provoquées par la crise sanitaire sur les circuits d'approvisionnement et de distribution de médicaments, l'agence nationale de sécurité du médicament et des produits de santé (ANSM) a recensé, en 2020, 2 446 signalements de ruptures et risques de rupture de stock, contre 1 504 en 2019, soit une augmentation de près de 63 % 398 ( * ) . Or l'année 2019 correspondait déjà à une progression du nombre de signalements de 73 % par rapport à 2018 (871 signalements).
Afin de faire face aux tensions d'approvisionnement, au début de la crise sanitaire, sur cinq médicaments indispensables pour les patients hospitalisés en réanimation - le cisatracurium, l'atracurium, le rocuronium, la kétamine et le midazolam -, un dispositif de régulation nationale a été mis en place, mobilisant notamment un réseau de six pharmacies à usage intérieur (PUI) et l'établissement pharmaceutique de l'AP-HP. Ces quelques structures hospitalières, les seules en capacité de réaliser des préparations à partir de matières premières, se sont ainsi attelées à la production de curares.
À titre d'exemple, un partenariat entre l'agence générale des équipements et produits de santé (Ageps) de l'AP-HP - dont dépend l'établissement pharmaceutique de l'AP-HP -, un établissement pharmaceutique privé, l'ANSM et les centres hospitaliers universitaires (CHU) de Lille et Lyon a permis la production, pour le compte de l'Ageps, d'environ 200 000 ampoules de cisatracurium.
Afin de pérenniser cette démarche, le b du 2° du I de l'article 35 du PLFSS pour 2022 modifie l'article L. 5121-1 du code de la santé publique afin d'introduire, au sein de la catégorie des préparations hospitalières, le statut de préparations hospitalières spéciales. Définies par décret en Conseil d'État, ces préparations ont vocation, en fonction des difficultés techniques de leur fabrication ou de la faible disponibilité des substances actives nécessaires, à être réalisées par des structures habilitées, dans des conditions précisées par ce même décret, par le ministre de la santé. La réalisation des préparations hospitalières spéciales pourra, le cas échéant, être confiée par ces structures, sous leur responsabilité, en sous-traitance, à un établissement pharmaceutique autorisé pour la fabrication de médicaments 399 ( * ) .
Les structures habilitées à réaliser ces préparations pourront être :
- des PUI ;
- des établissements pharmaceutiques des établissements de santé ;
- l'établissement pharmaceutique de l'agence nationale de santé publique, plus communément dénommée Santé publique France.
L'autorisation pour préparer ces préparations hospitalières spéciales sera délivrée à titre exceptionnel et temporaire par l'une des deux autorités suivantes, suivant le contexte :
- soit le directeur général de l'ANSM en cas de rupture de stock d'un médicament d'intérêt thérapeutique majeur (MITM) ;
- soit le ministre de la santé pour faire face à une menace ou une crise sanitaire grave.
Le II de l'article 35 du PLFSS pour 2022 complète l'article L. 5121-21 du code de la santé publique afin d'y préciser que, pour l'application des dispositions de l'article L. 5121-1 du même code, relatives aux caractéristiques des spécialités, produits et préparations devant être regardés comme des médicaments humains, les hôpitaux des armées sont assimilés à des établissements de santé et la pharmacie centrale des armées pourra être habilitée à réaliser des préparations hospitalières spéciales.
L'impact financier annuel de la mesure pour l'assurance maladie est estimé, par l'étude d'impact annexée au PLFSS, à 1,5 millions d'euros.
B. L'adaptation du cadre des préparations magistrales et hospitalières pour renforcer l'accès des patients aux traitements
Le 1° et le a du 2° du I de l'article 35 du PLFSS pour 2022 modifient, à l'article L. 5121-1 du code de la santé publique, les définitions, respectivement, des préparations magistrales et des préparations hospitalières afin de faciliter leur accès aux patients.
En l'état du droit vigueur, le recours à une préparation magistrale ne peut être envisagé qu'en l'absence de spécialité autorisée 400 ( * ) dans l'indication considérée. Le 1° du I de l'article 35 du PLFSS pour 2022 complète les hypothèses dans lesquelles le recours à une préparation magistrale pourrait être justifié, afin de tenir compte des situations suivantes :
- l'absence de spécialité pharmaceutique adaptée : une spécialité peut être disponible mais peut se révéler inadaptée pour le traitement d'un patient dans le cadre d'une prise en charge relevant de la médecine personnalisée. C'est notamment le cas en matière de transplantation de microbiote fécal. Bien que les autorisations de spécialités à base de microbiote fécal aient vocation à se multiplier, l'étude d'impact annexée au PLFSS insiste sur la nécessité de conserver aux établissements de santé la possibilité de réaliser et délivrer des préparations magistrales mieux adaptées aux besoins des patients ;
- l'absence de commercialisation effective : bien que théoriquement disponible, une spécialité peut ne pas faire l'objet d'une commercialisation effective, faute de commercialisation par l'entreprise exploitante ou de demande d'inscription au remboursement.
En conséquence, il est prévu que le recours à une préparation magistrale soit possible « lorsqu'il n'existe pas de spécialité pharmaceutique adaptée ou disponible, y compris du fait de l'absence de commercialisation effective ».
Les mêmes hypothèses sont retenues par le a du 2° du I de l'article 35 du PLFSS pour 2022 pour le recours aux préparations hospitalières. En l'état du droit en vigueur, celles-ci peuvent être accessibles en l'absence de spécialité pharmaceutique disponible ou adaptée, mais l'hypothèse de l'absence de commercialisation effective n'était pas encore explicitement envisagée.
L'impact financier annuel de la mesure pour l'assurance maladie est estimé, par l'étude d'impact annexée au PLFSS, à 14 millions d'euros.
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté, en première lecture, un amendement rédactionnel du rapporteur général.
III - La position de la commission
La commission se félicite de la transposition en France d'un modèle qui a d'ores et déjà fait ses preuves aux États-Unis pour la production de médicaments essentiels régulièrement exposés à des risques de tension d'approvisionnement. En septembre 2018, a en effet été lancée l'initiative Civica Rx , entreprise sans but lucratif, réunissant près de 120 organisations de santé rassemblant près du tiers des hôpitaux américains. Dans le cadre de cette initiative, l'entreprise s'engage à rendre disponibles et à des prix abordables et transparents des spécialités génériques essentielles, notamment par la passation de commandes et de contrats avec de long terme avec des sous-traitants.
Le Gouvernement justifie, dans l'étude d'impact annexée au PLFSS, l'insertion en loi de financement de sécurité sociale de dispositions ayant trait au circuit de production et d'approvisionnement des médicaments par le fait que « la mesure aura un impact sur les dépenses de l'assurance maladie par le biais du financement par dotation pour mission d'intérêt général qui sera versé aux établissements de santé concernés. »
Pour autant, l'article 35 du PLFSS ne modifie pas l'article L. 162-22-13 du code de la sécurité sociale instituant la dotation annuelle de financement des missions d'intérêt général et d'aide à la contractualisation (Migac) des établissements de santé en médecine, chirurgie et obstétrique (MCO). La commission a donc adopté un amendement n° 177 visant à préciser que les Migac auront vocation à financer les activités des établissements en MCO dûment habilités à produire des préparations hospitalières spéciales. Pour mémoire, dans un rapport 401 ( * ) de 2014 sur la recevabilité financière des amendements et propositions de loi, l'ancien président de la commission des finances du Sénat, M. Philippe Marini, rappelait que l'intention du Gouvernement exprimée dans l'étude d'impact peut servir de base de comparaison afin de valider la recevabilité d'un amendement au titre de l'article 40 de la Constitution.
Par ailleurs, afin d'étendre le champ des sous-traitants susceptibles d'être mobilisés par les établissements pharmaceutiques publics pour la réalisation de préparations hospitalières spéciales, la commission a adopté un amendement n° 176 ouvrant la possibilité à ces établissements de confier la production, en tout ou partie, de ces préparations à des pharmacies d'officine sous-traitantes dûment autorisées par l'ARS.
En effet, il existe aujourd'hui un réseau de pharmacies d'officine autorisées par le directeur général de l'ARS compétente à exercer une activité de sous-traitance pour l'exécution de préparations au profit d'autres officines. À titre d'exemple, ces pharmacies se sont mobilisées de façon réactive, en 2009, pour la production de solutions buvables de Tamiflu® et, en 2019, pour la production de corticoïdes.
Enfin, la commission a adopté un amendement rédactionnel n° 175.
La commission vous demande d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article 36
Accès
direct au marché remboursé post avis HAS
Cet article propose de créer un nouveau régime d' « accès direct » permettant la prise en charge à l'issue de l'avis de la Haute Autorité de santé, sans attendre la fixation du prix
Formulant certaines réserves sur la bonne articulation du nouveau dispositif avec l'accès précoce récemment mis en oeuvre, la commission vous demande d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - Le dispositif proposé
A. Un dispositif complémentaire de l'accès précoce pour réduire les délais d''accès aux médicaments innovants
1. Une lacune constatée dans le délai d'accès à l'innovation
À l'issue de l'obtention d'une autorisation de mise sur le marché, un médicament doit, pour être pris en charge par l'assurance maladie, être inscrit sur les listes de remboursements. À l'issue de son évaluation par la Haute Autorité de santé (HAS), le médicament fait l'objet de négociations tarifaires avec le Comité économique des produits de santé (CEPS).
Si la durée de cette procédure d'accès au marché remboursé après une demande d'inscription ne peut réglementairement excéder 180 jours 402 ( * ) , celle-ci peut fortement varier et excéder largement les délais moyens de 147 jours pour les premiers demandes sur la liste en sus et 144 jours pour les premières demandes en ville.
Face à ces délais qui retardent l'accès des patients aux médicaments innovants, le dispositif d'« accès précoce » adopté en LFSS pour 2021 403 ( * ) permet une prise en charge d'un médicament présumé innovant jusqu'à trois ans avant l'obtention d'une autorisation de mise sur le marché .
Cependant, la prise en charge au titre de l'accès précoce n'est ouverte que pour les maladies graves, rares et invalidantes et face à un besoin thérapeutique.
Sur la base des avis rendus par la Commission de la Transparence en 2020, une vingtaine d'indications présentent des évaluations apparaissant compatibles avec cette mesure d'accès direct tout en n'étant pas disponible au travers du système d'accès précoce.
Cette indisponibilité en accès précoce découle essentiellement de non-sollicitation de la part des laboratoires exploitants et n'est, dès lors, pas liée à une inéligibilité de l'indication, plus de la moitié des indications hors accès précoce y étant pourtant éligibles à la vue des critères d'entrée.
Source : Réponse de la direction de la sécurité sociale au questionnaire de la rapporteure
2. La mise en oeuvre d'une mesure du plan Innovation 2030
En conclusion du Conseil stratégique des industries de santé (CSIS) 2021 , la mesure 4 « Permettre une équité d'accès aux soins pour les patients et offrir aux innovations un cadre d'accès au marché accéléré et simplifié » du plan « Innovation santé 2030 » a prévu au titre de ses déclinaisons « la mise en place d'un mécanisme d'accès immédiat au marché avec une ASMR 1 à 4 post-avis de la Haute autorité de santé, comparable au système allemand d'accès au marché, avec un test pendant 2 ans » .
L'évaluation préalable jointe au PLFSS envisage, du fait d'une prise en charge anticipée de six mois à un an des médicaments entrant dans ce dispositif, un surcoût pour l'assurance maladie de l'ordre de l'ordre 100 millions d'euros chaque année.
B. Un nouvel accès dérogatoire à titre expérimental
1. Un nouveau dispositif de prise en charge...
Le présent article entend traduire cette mesure légalement à travers l'expérimentation d'un nouveau dispositif dérogatoire et permettre un accès anticipé prioritairement destiné aux médicaments non éligibles à l'accès précoce mais présentant un service médical rendu important .
Le choix d'une expérimentation
Une expérimentation a été préférée dans la mesure où (1) le besoin nécessitait d'être validé au regard de la réforme récente de l'accès précoce et (2) certains risques ont été identifiés qui nécessitaient de passer par une phase expérimentale , à savoir
(i) l'articulation avec l'accès précoce et l'effet contreproductif possible de retarder l'accès aux patients pour des médicaments qui répondent à un besoin non couvert ;
(ii) une négociation complexifiée du fait du type de produits (probablement ASMR IV avec comparateurs) mais d'un accès patients préalable et donc la volonté de tester notamment le système de prise en charge (comme accès précoce mais avec remises plus élevées) et le passage par une décision du comité économique des produits de santé en cas d'absence d'accord au bout des douze mois d'accès direct.
Source : Réponse de la direction de la sécurité sociale au questionnaire de la rapporteure.
Le I du présent article prévoit ainsi « à titre expérimental » un dispositif dit d'« accès direct » pour des spécialités pharmaceutiques ne faisant pas l'objet d'une autorisation d'accès précoce .
Ce dispositif ouvre la possibilité d'une prise en charge de cette spécialité par l'assurance maladie pour une indication donnée et sous réserve d'une autorisation de mise sur le marché pour cette indication.
Ce dispositif est réservé aux spécialités ne faisant pas l'objet d'une inscription sur les listes de remboursements de droit commun (article L. 162-17 du code de la sécurité sociale et article L. 5123-2 du code de la santé publique pour les établissements hospitaliers) et n'étant pas pris en charge au titre d'un accès précoce (article L. 162-16-5-1 du code de la sécurité sociale) ou d'un accès compassionnel (article L. 162-16-5-2 du même code).
Cette prise en charge n'est ouverte que pour une durée maximale d'un an et est réservée à certains établissements de santé ou établissements disposant d'une pharmacie à usage intérieur (PUI).
2. ... Conditionné à la satisfaction à différents critères
Le II précise l'encadrement de ce nouveau dispositif dérogatoire en prévoyant cinq conditions à satisfaire pour le bénéfice de la prise en charge au titre de l'accès direct, celle-ci étant accordée ensuite par arrêté des ministres de la santé et de la sécurité sociale .
Le 1° du II encadre les conditions de la demande de prise en charge au titre de l'accès direct. Celle-ci doit ainsi être formulée au plus tard un mois à l'issue de l'avis rendu par la commission de la transparence de la Haute Autorité de santé sur la demande d'inscription sur une liste de remboursements.
Par ailleurs, ce même 1° borne la durée de l'expérimentation en prévoyant un dépôt de demande au plus tard deux ans après son début . Si la date de démarrage de l'expérimentation est renvoyée à un décret, elle devra intervenir avant le 1er juillet 2022 .
Le 2° précise, dans les cas de médicaments réservés à l'usage hospitalier, la nécessité de répondre aux critères d'inscription sur la liste en sus pour les indications concernées. Par ailleurs, une demande d'inscription sur cette même liste doit être conjointement formulée.
Le 3° fixe un critère d'efficacité minimal de la spécialité pour assurer son éligibilité au dispositif, exigeant un service médical rendu (SMR), apprécié par la HAS, supérieur à un niveau fixé par décret . Si ce niveau n'est pas précisé dans l'évaluation préalable, le dispositif a vocation à concerner des médicaments avec un SMR important .
Le 4° complète ces exigences avec un niveau minimal d'amélioration du service médical rendu (ASMR). L'évaluation préalable indique un niveau envisagé d'ASMR 1 à 4 , soit au moins mineure .
Enfin, le 5° prévoit un engagement de l'exploitant chargé de garantir la continuité des traitements initiés pendant la durée de l'accès direct et, à l'issue de la prise en charge liée, pour une durée complémentaire d'un an . Cet engagement de continuité est levé en cas d'arrêt de commercialisation pour une raison sérieuse relative à la sécurité des patients.
En cas de manquement à cette dernière obligation , le B du IX du présent article prévoit, après mise en demeure, la possibilité d'une sanction financière prononcée par les ministres précités. Celle-ci peut s'élever à 30 % du chiffre d'affaires réalisé sur la spécialité durant les deux dernières années. Elle est versée à la caisse nationale d'assurance maladie.
3. Des conditions de prise en charge définies
Aux termes du IV de l'article, l'autorisation d'accès direct emporte une série de conséquences :
- l'interdiction d'inscription de la spécialité sur une liste de remboursements pour une autre indication ( 4° ) ;
- la dispense d'inscription sur la liste en sus pour les achats hospitaliers ( 5° ) ou le caractère réputé satisfait d'inscription sur la liste des médicaments autorisés à être vendus par une pharmacie à usage intérieur ( 6° ).
Par ailleurs, concernant les conditions directes de prise en charge :
- le prescripteur indique sur l'ordonnance la mention de prise en charge au titre de l'accès direct et informe le patient des conditions de prise en charge ( 7° ) ;
- la spécialité est prise en charge en sus des prestations d'hospitalisation ( 8° ).
Enfin, les dispositions de droit commun relatives au respect des règles de tarification, au service du contrôle médical ou à l'inscription des indications et les sanctions applicables en cas de non-respect sont applicables ( 9° et 10° ).
C. Une sortie organisée du dispositif
1. Une durée maximale d'un an
Aux termes du V du présent article, la prise en charge prend fin au titre de chaque indication donnée au plus tard un an après la date de décision de prise en charge.
Cependant, la prise en charge peut expirer avant cette date :
- en cas d'inscription sur une liste de remboursements et de publication du prix ou du tarif de responsabilité ( 1° ) ;
- en cas de demande de l'exploitant ( 2° ) ;
- sur décision ministérielle , en cas de refus d'inscription sur l'une des listes de remboursements ou de retrait de la demande d'inscription sur l'une de ces listes ( 3° ).
2. Deux régimes de prise en charge prévus
À l'issue de la prise en charge au titre de l'accès direct , le A du IX prévoit différents cas de figure :
- dans le cas d'une inscription sur l'une des listes de remboursements, alors la bascule dans le droit commun s'opère et les conditions de dispensation et de prise en charge sont celles de ces listes ( 1° ) ;
- dans le cas d'une non-inscription sur les listes, les dernières conditions de dispensation s'appliquent.
Enfin, par dérogation, les médicaments peuvent être achetés par les hôpitaux sans figurer sur la liste prévue à l'article L. 5123-2 du code de la santé publique et s'ils ne sont pas réservés à l'usage hospitalier, ils sont réputés inscrits sur la liste des médicaments autorisés à être vendus par une pharmacie à usage intérieur.
D. Un système propre de tarification et de remises
1. Une tarification propre
Si le Gouvernement précise que le prix est librement fixé par l'industriel, le III prévoit un nouveau système de « compensation » accordée à l'entreprise exploitant la spécialité, qui s'apparente à un tarif.
Le deuxième alinéa du III précise que son montant est modulé selon la catégorie de spécialité , la nature de celle-ci, la taille de la population cible, l'existence de comparateurs et enfin le niveau d'ASMR . Les catégories et les montants par catégories sont fixés par arrêté.
2. Des remises applicables durant l'accès direct
Le IV organise un système de remises comparable aux autres remises applicables aux médicaments.
Ainsi, le 1° prévoit une déclaration par l'exploitant au CEPS du montant de l'indemnité maximale facturée aux établissements de santé pour la spécialité et, aux termes du 2° , une communication le 15 février de chaque année au CEPS du chiffre d'affaires de l'année précédente réalisé sur cette spécialité pour chacune des indications, ainsi que du nombre d'unités fournies.
Le 3° prévoit le versement annuel aux organismes d'assurance maladie de remises . Celles-ci correspondent à la différence entre le chiffre d'affaires déclaré et celui qui aurait résulté de la vente des mêmes doses au tarif de « compensation » , ce pour la période et l'indication concernée. Le même 3° précise les conditions de calcul du chiffre d'affaires retenu.
Le VIII prévoit enfin l'application aux médicaments pris en charge au titre de l'accès direct, au sein de l'article L. 162-18, des dispositions conditionnant le remboursement par l'assurance maladie au versement obligatoire de remises par les entreprises exploitantes, importatrices ou distributrices.
3. Un nouveau système de remises post accès direct
a) Un prix net de référence fixé à l'issue de l'accès direct
Le C du VI détermine les conditions d'application des conventions relatives aux spécialités ayant bénéficié de l'accès direct et les conditions de fixation du prix net de référence ainsi que l'intégration dans son calcul des remises de droit commun.
b) Des remises spéciales lors de la bascule dans le droit commun
À l'issue de l'accès direct et en cas d'inscription sur une liste de remboursements, le VI prévoit un nouveau mécanisme de remises supplémentaires prévues au sein de la convention ou de la décision fixant le prix de référence de la spécialité ayant fait l'objet d'une prise en charge au titre de l'accès direct pour une indication.
Le CEPS est ainsi chargé de calculer :
- le chiffre d'affaires qui aurait résulté de la vente au prix net de référence des unités vendues sur l'ensemble de la période (1° du A) ;
- le chiffre d'affaires facturé aux établissements après déduction de la remise prévue au IV, sur la même période (2° du A).
Aussi, dans le cas où le chiffre d'affaires résultant de l'application du prix net de référence se trouvait être inférieur au chiffre d'affaires constaté durant la période d'accès direct, l'exploitant se trouverait dans l'obligation de verser une remise supplémentaire correspondant à la différence.
Une régulation de prix a posteriori est ainsi faite dans le cas de la fixation d'un tarif de remboursement inférieur au tarif pratiqué durant la période d'accès direct.
Le B du VI prévoit le versement de ces remises complémentaires en une seule fois , au titre de l'année d'inscription au remboursement.
c) Des remises maintenues an cas de non inscription
Aux termes du VII de l'article, le s dispositions relatives aux remises figurant aux A et B du VI demeurent applicables lorsque la prise en charge cesse sans relais par un remboursement pour l'indication donnée.
Le CEPS peut alors retenir un prix de référence selon les critères de droit commun de fixation et de modification des prix.
F. Modalités d'application et d'évaluation
Le X prévoit un décret en Conseil d'État chargé de définir les modalités d'application du présent article.
Enfin, l'expérimentation à cet article doit faire l'objet d'une évaluation dans un délai de vingt-et-un mois après son démarrage ( XI ), soit au plus tard en avril 2024, celle-ci étant adressée au Parlement.
Si son contenu doit être précisé par décret, l'évaluation préalable indique que le rapport d'évaluation présentera les demandes déposées pour bénéficier du dispositif et leurs caractéristiques (évaluations de la HAS relatives aux comparateurs, à l'amélioration du service médical rendu ainsi que leur population cible, les aires thérapeutiques visées et leur gravité ou rareté le cas échéant), l'évolution parallèle du nombre de dépôts de demandes d'autorisations d'accès précoce et leur temporalité, la durée des accès directs, le nombre de patients traités pendant cet accès anticipé, les dépenses engagées au titre de cet accès, le passage ou non dans le remboursement de droit commun, les modalités et les conditions tarifaires de cet éventuel passage dans le droit commun, notamment au regard de spécialités comparables n'ayant pas bénéficié du dispositif.
II - Les modifications adoptées par l'Assemblée nationale
Outre une série de modifications rédactionnelles, différentes modifications substantielles ont été adoptées par l'Assemblée nationale à l'initiative du Gouvernement et du rapporteur général de la commission des affaires sociales.
A. Un articulation renforcée avec le dispositif d'accès précoce
À l'initiative du Gouvernement, l'article a été modifié en vue d'aligner le régime expérimental d'accès direct sur le modèle de l'accès précoce et d'en assurer une meilleure articulation.
Ainsi, le champ défini au I a été précisé et sont désormais éligibles à l'accès direct les spécialités inscrites sur la liste hospitalière pour d'autres indications ou, dans ce même cas, les spécialités faisant l'objet d'une prise en charge au titre de l'accès précoce . Seuls sont donc exclus les médicaments disponibles en ville, le Gouvernement rappelant que les remises ne sont aujourd'hui pas appelées sur les facturations en ville.
Principale modification, un 1° bis nouveau a été introduit au II de l'article qui prévoit que, pour bénéficier du régime d'accès précoce, la spécialité doit satisfaire à une condition supplémentaire. L'exploitant doit ainsi, sur les indications considérées, avoir au plus tard au moment de sa demande d'AMM, déposé une demande d'autorisation d'accès précoce. La HAS doit par surcroît s'être prononcée sur cette demande et l'exploitant l'avoir maintenue et, dans le cas d'une décision d'autorisation, ne pas avoir demandé le retrait de cette dernière.
B. Une révision des dispositions relatives aux remises et à la fixation du prix
Par ailleurs, l'amendement gouvernemental a précisé les conditions de déclaration au CEPS de l'indemnité maximale demandée, prévoyant une dispense lorsqu'un prix maximal de vente est déjà défini ou une prise en charge pour une indication est déjà prévue.
Surtout, par le V bis créé, le Gouvernement a entendu préciser qu'à défaut d'un accord conventionnel entre l'exploitant et le CEPS dans les dix mois après l'ouverture de l'accès direct, le prix de vente au public, le prix de cession, ou le tarif de responsabilité et prix limite de vente aux établissements seraient fixés par décision du CEPS, et ce avant la fin du douzième mois. Ce délai correspond ainsi à la fin de la durée de prise en charge au titre de l'accès direct , afin d'assurer la bascule vers le droit commun.
Ont enfin été complétées les dispositions du VI afin de prévoir une restitution au laboratoire dans le cas d'un prix de remboursement finalement supérieur au prix facturé aux établissements de santé déduit des remises .
Parallèlement, à l'initiative du rapporteur général, l'Assemblée nationale a prévu :
- la suppression du mécanisme de compensation au bénéfice du système de remises, avec l'application aux remises accordées d'un barème progressif par tranche de chiffre d'affaires , sur le modèle des remises applicables dans le cadre de l'accès précoce. L'intention est celle d'un barème cependant plus strict, du fait des alternatives existantes pour ces spécialités ou de leurs évaluations inférieures.
- un plafonnement du prix d'achat des spécialités dans la période de continuité du traitement à l'issue de la prise en charge au titre de l'accès direct, à défaut d'une prise en charge de droit commun.
L'Assemblée nationale a adopté cet article ainsi modifié.
III - La position de la commission
A. Une intention partagée de soutien à un accès rapide à l'innovation
La rapporteure soutient l'initiative du Gouvernement de conduire une expérimentation d'un nouveau dispositif d'accès anticipé à l'innovation, qui semble par ailleurs susciter l'intérêt des industriels .
Cet article, qui traduit une mesure du CSIS 2021, s'inscrit également dans la ligne des travaux de la commission et notamment du rapport sur l'innovation en santé publié en amont des conclusions du CSIS 2021 par les sénatrices Annie Delmont-Koropoulis et Véronique Guillotin. La commission avait alors constaté l'importance de la question des délais d'accès à l'innovation pour les patients et les retards que pouvait occasionner le processus de fixation des prix après l'avis de la HAS.
B. Plusieurs réserves et points de vigilance
Si elle partage l'esprit du dispositif, la rapporteure, au cours de ses travaux préparatoires à l'examen du PLFSS, a constaté plusieurs aspects pouvant justifier des ajustements.
Sur le champ du dispositif, la rapporteure a été notamment sensibilisée sur l'enjeu que pouvait représenter, en termes d'attractivité du dispositif, l'intégration des extensions d'indications.
• Concernant l'articulation avec l'accès précoce , la rapporteure souhaite que le dispositif d'accès direct n'affaiblisse pas le système d'accès dérogatoire adopté en LFSS pour 2021. Il s'agit de préserver le dispositif d'accès précoce et, particulièrement, l'accès précoce pré-AMM .
À ce titre, elle souscrit aux modifications apportées à l'Assemblée nationale pour éviter un déport des médicaments éligibles à l'accès précoce . Particulièrement sensible à ce sujet de préoccupation, la rapporteure souligne que l'accès direct doit bien être conçu et mis en oeuvre comme un complément à l'accès précoce, alors que son champ et les critères d'éligibilité envisagés sont bien plus larges . Il ne doit pas être plus incitatif que l'accès précoce et engager les laboratoires à attendre l'autorisation de mise sur le marché et, partant, retarder l'accès des patients à l'innovation.
La rapporteure souligne également que l'encadrement de ce nouveau dispositif et les éventuelles demandes supplémentaires nécessiteront des moyens supplémentaires pour la Haute Autorité de santé , non précisés pour le moment.
La rapporteure s'interroge en outre, alertée sur ce point par les industriels, sur la compatibilité du calendrier proposé avec les délais nécessaires au dépôt des demandes d'autorisation de mise sur le marché et d'autorisation d'accès précoce. Par ailleurs, interpellée sur les délais parfois longs des décisions administratives, la commission a adopté l'amendement n° 180 visant à prévoir la publication de l'arrêté de prise en charge dans un délai de six semaines au plus .
Enfin, la rapporteure constate que tous les médicaments visés par le dispositif d'accès direct n'ont pas vocation à être éligibles à l'accès précoce . Il conviendrait ainsi de ne prévoir le dépôt de la demande d'accès précoce qu'en cas de correspondance des critères. La commission a ainsi adopté l'amendement n° 178 limitant le dépôt nécessaire d'une demande d'accès précoce aux spécialités qui y sont éligibles .
Concernant le mécanisme de remises prévues, la rapporteure a constaté les évolutions opérées à l'Assemblée nationale. Elle souligne son souci, pour que le dispositif soit attractif, d'une bonne lisibilité des conditions de prise en charge . À ce titre, si l'application d'un système comparable à l'accès précoce peut sembler pertinente, la rapporteure estime cependant que les deux dispositifs doivent être bien distingués et préserver un avantage au dispositif d'accès précoce, en cela que l'apport des médicaments éligibles à ce dernier a vocation à être supérieur.
• Le Gouvernement a enfin indiqué à votre rapporteure que cet article pourrait faire l'objet de nouvelles modifications , à son initiative, lors de son examen en première lecture au Sénat.
Enfin, la commission a adopté les amendements rédactionnels n° 179 et n° 181.
La commission vous demande d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article 36 bis
(nouveau)
Rapport visant à présenter l'avancement de la
révision
des actes hors nomenclature et de leur financement
Cet article, inséré par l'Assemblée nationale, vise à demander la remise d'un rapport du Gouvernement au Parlement sur l'avancement de la réforme des actes hors nomenclature et leur financement.
La commission vous demande de supprimer cet article.
I - Le dispositif proposé
A. Les actes innovants hors nomenclature
La direction générale de l'offre de soins (DGOS) a mis en place en 2015 le référentiel des actes innovants hors nomenclature (RIHN). Les actes innovants de biologie médicale et d'anatomocytopathologie compris dans ce référentiel peuvent ainsi faire l'objet d'une prise en charge précoce avant même leur inscription aux nomenclatures 404 ( * ) .
Ce dispositif de soutien à l'innovation en santé est financé par une enveloppe globale dévolue au RIHN. Face à une demande croissante d'inscription au RIHN et une enveloppe budgétaire limitée, le financement des actes connaît de fortes tensions reconnues par le Gouvernement 405 ( * ) . Celui-ci a annoncé en 2019 l'engagement des travaux par la DGOS, l'assurance maladie et la Haute Autorité de santé pour permettre une inscription à la nomenclature de certains actes de routine de biologie médicale.
Plus récemment, le Gouvernement a lancé le conseil stratégique des industries de santé 2021 (CSIS) avec comme objectif de renforcer l'innovation en santé au stade de la recherche, du financement, de l'accès au marché et de l'industrialisation.
B. Le dispositif proposé
Le présent article introduit à l'Assemblée nationale par un amendement de Michel Lauzzana (La République en Marche), sous-amendé par le Gouvernement, prévoit la remise d'un rapport du Gouvernement au Parlement dans un délai d'un an à compter de la promulgation de la présente loi.
Ce rapport présenterait l'avancement de la révision des actes hors nomenclature et de leur financement et examinerait spécifiquement les possibilités de création d'une enveloppe pour assurer la prise en charge des actes de médecine génomique.
II - La position de la commission
La rapporteure regrette que la révision des actes hors nomenclature n'aboutisse pas en dépit des annonces régulières du Gouvernement . Cette révision devrait pourtant être une priorité. La mission d'information de la commission sur l'innovation en santé avait constaté « les conséquences néfastes [du RIHN] sur la prise en charge des patients, en particulier ceux atteints de cancer en les privant des tests dits “compagnons” essentiels pour préciser le diagnostic et accompagner le développement de la médecine personnalisée en oncologie ». Elle avait donc recommandé de revoir l'enveloppe RIHN et de redéfinir les cotations de ces actes 406 ( * ) .
Toutefois, quelle que soit l'importance du sujet de l'innovation en santé et suivant sa position habituelle sur les demandes de rapport adressées au Gouvernement, la commission a adopté un amendement n° 183 de sa rapporteure de suppression de l'article.
La commission vous demande de supprimer cet article.
Article 37
Recours
aux médicaments biosimilaires
Cet article propose de rétablir les dispositions du code de la santé publique permettant la dispensation par les pharmaciens de médicaments biosimilaires en substitution au produit prescrit.
La commission vous demande d'adopter cet article modifié par les amendements qu'elle a adoptés.
I - Le dispositif proposé
A. Une nouvelle tentative de dispositif relatif à la substitution de biosimilaires
1. Un potentiel d'économies pour l'assurance maladie
Le Gouvernement indique dans l'évaluation préalable de l'article 37 que les médicaments biologiques concernés par des biosimilaires représentent plus de 2,5 milliards d'euros de dépenses annuelles en 2021-2022.
Surtout, les échéances de brevet pourraient augmenter le panier de soins biosimilarisés d'1,3 milliard d'euros à l'horizon 2025.
Un médicament biologique est une substance produite à partir d'une cellule ou d'un organisme vivant, ou dérivée de ceux-ci. Les vaccins, les anticorps monoclonaux ou les facteurs de croissance sont des exemples de produits biologiques.
La production des médicaments biologiques est complexe car elle s'appuie sur des cellules ou des organismes vivants. En raison de la variabilité biologique de ces sources de production, des différences de fabrication sont inévitables et elles peuvent impacter les propriétés cliniques des produits. Un médicament biosimilaire est similaire à un médicament biologique dit de référence car déjà autorisé en Europe.
Tout médicament biologique dont le brevet est tombé dans le domaine public peut être copié. Cette copie est désignée comme biosimilaire. Les produits biosimilaires ne pouvant être strictement identiques au produit de référence, le principe de substitution, valable pour les médicaments chimiques et les génériques qui sont leurs copies, ne peut donc pas s'appliquer automatiquement.
Source : Agence nationale de sécurité du médicament 407 ( * )
Le code de la santé publique prévoit deux définitions respectivement des médicaments biologiques et des médicaments biologiques similaires.
Article L. 5121-1 du code de la santé publique
14° Médicament biologique, tout médicament dont la substance active est produite à partir d'une source biologique ou en est extraite et dont la caractérisation et la détermination de la qualité nécessitent une combinaison d'essais physiques, chimiques et biologiques ainsi que la connaissance de son procédé de fabrication et de son contrôle ;
15° a) Sans préjudice des articles L. 611-2 et suivants du code de la propriété intellectuelle, médicament biologique similaire, tout médicament biologique de même composition qualitative et quantitative en substance active et de même forme pharmaceutique qu'un médicament biologique de référence mais qui ne remplit pas les conditions prévues au a) du 5° du présent article pour être regardé comme une spécialité générique en raison de différences liées notamment à la variabilité de la matière première ou aux procédés de fabrication et nécessitant que soient produites des données précliniques et cliniques supplémentaires dans des conditions déterminées par voie réglementaire ;
Un médicament biologique ne peut être qualifié de médicament biologique de référence que si son autorisation a été délivrée au vu d'un dossier comportant, dans des conditions fixées par voie réglementaire, l'ensemble des données nécessaires et suffisantes à elles seules pour son évaluation ;
b) Groupe biologique similaire, le regroupement d'un médicament biologique de référence et de ses médicaments biologiques similaires , tels que définis au a) du présent 15°. Ils sont regroupés au sein de la liste de référence des groupes biologiques similaires établie par l'Agence nationale de sécurité du médicament et des produits de santé.
Alors que le prix des médicaments biosimilaires est inférieur de 15 à 30 % aux médicaments biologiques de référence , ils représentent un potentiel non négligeable d'économies pour l'assurance maladie.
Plusieurs mesures incitatives ont déjà été adoptées en vue de favoriser la pénétration des biosimilaires concernant :
- l'incitation à l'utilisation en intra-hospitalier ;
- les prescriptions hospitalières exécutées en ville ;
- l'utilisation dans le domaine ambulatoire.
Le présent article entend compléter ces mesures en rétablissant une possibilité de substitution par les pharmaciens d'officine. L'objectif du Gouvernement est de favoriser la pénétration sur le périmètre des produits de ville, qu'il estime inférieure de 10 points à la cible des 80 %.
Seuls deux biosimilaires devraient au départ être rendus substituables, la somatropine et le pegfilgrastim. Ces deux molécules pourraient seules générer un gain de 6 millions d'euros par an en atteignant la cible des 80 %. L'étude d'impact prévoit des économies progressives, avec un potentiel d'économie nouvelle de 20 millions d'euros indiqué pour 2025.
2. Une possibilité de substitution inscrite en LFSS pour 2014 et abrogée en 2020
La loi de financement de la sécurité sociale avait inscrit au sein de des articles L. 5125-23-2 et L. 5125-23-3 du code de la santé publique une possibilité de substitution par les pharmaciens pour les médicaments biologiques similaires . Cette substitution n'était alors ouverte qu'en première délivrance ou en vue de pallier une difficulté de poursuite de traitement.
Le décret en Conseil d'État permettant l'application de cet article n'a cependant jamais été pris et le Gouvernement a proposé dans le projet de loi de financement pour 2020 l'abrogation des dispositions relatives à la substitution de biosimilaires 408 ( * ) .
Faute de disposition autorisant la substitution, l'abrogation réalisée en 2020 conduit à un retour au droit commun , c'est-à-dire aux règles de l'article L. 5125-3 du code de la santé publique et l'impossibilité pour le pharmacien d'officine de dispenser un autre médicament que celui qui a été prescrit sans l'accord exprès et préalable du médecin.
B. Un quasi rétablissement de dispositions abrogées en 2020
1. Un rétablissement des possibilités de substitution par les pharmaciens
Le I rétablit un article L. 5125-23-2 au sein du code de la santé publique, qui comme le précise le premier alinéa, déroge aux dispositions de l'article L. 5125-23 relatives à la délivrance des médicaments prescrits en vue de permettre la délivrance d'un médicament biologique similaire en substitution du médicament biologique prescrit .
Aux termes du même alinéa, cette substitution doit, pour être permise, satisfaire à différentes conditions nécessaires, dont trois figuraient déjà dans la rédaction de 2014 :
- le biosimilaire doit appartenir au même groupe biologique que le médicament biologique prescrit ( 1° ) ;
- le prescripteur n'a pas exclu la possibilité de cette substitution ( 4° ) ;
- les conditions de prise en charge par l'assurance maladie sont respectées ( 5° ).
En outre, la rédaction reprend les dispositions générales relatives aux modalités de délivrance en prévoyant :
- l'information du prescripteur lors de la substitution et l'inscription sur l'ordonnance du nom du biosimilaire ( alinéa 8 ) ;
- la délivrance d'un grand conditionnement dans le cas d'une prescription d'une durée de trois mois minimum ( alinéa 9 ).
Cependant, là où la rédaction de 2014 prévoyait que « la substitution est réalisée en initiation de traitement ou afin de permettre la continuité d'un traitement déjà initié avec le même médicament biologique similaire », la rédaction proposée au présent article ne précise pas de telles conditions et laisse donc la possibilité d'une substitution à tout moment durant le traitement .
Enfin, la rédaction proposée ne retient pas de décret nécessaire à la précision du dispositif et à son application .
2. Un encadrement renforcé des médicaments biologiques substituables
Nouveauté de la rédaction de 2021, le 2° prévoit comme autre critère nécessaire à la possibilité de substitution l'inscription du médicament sur une liste arrêtée par le ministre de la santé après avis de l'ANSM. Cette liste peut être assortie de conditions de substitution et d'information du prescripteur et du patient à l'occasion de cette substitution de nature à assurer la continuité du traitement avec le même médicament.
Par surcroît, le 3° prévoit comme autre critère nécessaire la satisfaction, lorsqu'elles existent, des conditions précitées.
3. Un plafonnement des remboursements au prix du biosimilaire le plus onéreux
Le 1° du II rétablit le V de l'article L. 162-16 du code de la sécurité sociale, abrogé en LFSS pour 2020, afin de prévoir que la délivrance d'un produit biosimilaire en substitution ne peut occasionner de dépense supplémentaire que la délivrance du biosimilaire du même groupe le plus cher . Dans la rédaction de 2014, le plafond retenu était celui du biosimilaire ou du générique le plus onéreux.
Il s'agit ici concrètement, dans le cas de la substitution d'un biosimilaire par le médicament biologique de référence, de plafonner le remboursement au biosimilaire le plus cher.
4. Un rôle et des objectifs fixés pour les pharmaciens
Le 2° du II modifie l'article L 162-16-1 du même code afin d'intégrer au sein de la convention entre l'union nationale des caisses d'assurance maladie (Uncam) et les syndicats de pharmaciens la participation des pharmaciens au développement des médicaments biosimilaires .
Le 3° du même II modifie enfin l'article L. 162-16-7 dudit code afin de prévoir au sein de l'accord entre l'Uncam et les syndicats de pharmaciens d'officine les objectifs de délivrance de médicaments biosimilaires sur le même modèle que les objectifs de délivrance fixés en matière de génériques.
II - Les modifications adoptées par l'Assemblée nationale
Lors de l'examen du projet de loi de financement, l'Assemblée nationale a adopté, outre une modification rédactionnelle, une série d'amendements sur cet article tendant à :
- encadrer les possibilités de mention « non substituable » et ainsi prévoir que l'exclusion de la possibilité de substitution par le prescripteur devait être formulée et justifiée expressément sur l'ordonnance, cette mention devant tenir à la situation du patient ;
- préciser l'information du patient par le pharmacien d'officine lorsque celui-ci procède à une substitution ;
- prévoir la remise d'un rapport ( III nouveau ) sur les dispositifs médicaux aux caractéristiques techniques et cliniques similaires pouvant faire l'objet d'une substitution.
L'Assemblée nationale a adopté cet article ainsi modifié.
III - La position de la commission
A. Des informations très lacunaires au moment de l'examen du présent texte
Dans les évaluations préalables jointes au PLFSS, le Gouvernement indique proposer ce dispositif eu égard aux positions nouvelles prises par l'Agence nationale de sécurité du médicament. Or, force est de constater qu'aucun avis nouveau ni rapport dédié n'a été publié par l'ANSM en 2021 et le dernier rapport de référence sur les biosimilaires demeure celui de 2016, antérieur à l'abrogation de l'ancien dispositif ...
Interrogée sur les informations nouvelles à disposition du Gouvernement, d'une part, lui permettant de proposer ce nouveau dispositif, mais aussi des parlementaires, d'autre part, chargés de l'approuver dans ce PLFSS, l'ANSM a répondu à votre rapporteur qu'une actualisation du rapport de 2016 faisait à cette date l'objet de consultations en cours des parties prenantes (associations de patients, professionnels de santé, industriels).
Cependant, l'Agence a signalé à votre rapporteure plusieurs éléments sommaires :
- il apparaît à l'ANSM, au regard de l'expérience européenne et des 65 biosimilaires autorisés, qu'une interchangeabilité 409 ( * ) est possible en primo-prescription comme en cours de traitement ;
- une substitution par le pharmacien serait possible mais, souligne l'agence, très progressive . Elle ne devrait concerner que des molécules bien connues et être strictement encadrée . En revanche, l'agence n'estime pas envisageable une substitution systématique sur le modèle des génériques .
Votre rapporteure ne peut que regretter que le Parlement ne dispose pas de l'ensemble des informations attendues de la part du Gouvernement et susceptibles d'éclairer son vote au moment de l'examen du texte en première lecture dans chacune des deux chambres. Le Gouvernement fait preuve ici d'une précipitation préjudiciable à la sincérité du texte.
B. Un rétablissement bienvenu de possibilités de substitution supprimées en 2020 contre l'avis de la commission 410 ( * )
Alors que la commission s'était opposée en 2019 à la suppression du dispositif issu de la LFSS pour 2014, la rapporteure soutient le rétablissement de ces dispositions, dont le potentiel d'économies est bienvenu pour l'assurance maladie .
La rapporteure souligne cependant, suivant en cela les recommandations de l'ANSM, que ces possibilités de substitution doivent être suffisamment encadrées et concerner des molécules offrant un recul suffisant .
Elle s'interroge par ailleurs sur la justification du décalage qui demeure entre le champ des molécules interchangeables et substituables.
La rapporteure émet cependant des réserves sur les précisions apportées sur l'information tant du prescripteur que du patient alors que, d'une part, le prescripteur n'est pas nécessairement joignable et que, d'autre part, l'information du patient relève du rôle systématique du pharmacien et ne nécessitait pas d'inscription dans la loi.
Enfin, pour intéressant que soit le sujet des dispositifs médicaux aux caractéristiques techniques et cliniques similaires, la commission est, de manière constante, opposée aux demandes de rapports au Parlement .
La commission a ainsi adopté un amendement n° 186 visant à supprimer la demande de rapport ainsi que deux amendements rédactionnels n° 184 et n° 185.
La commission vous demande d'adopter cet article modifié par les amendements qu'elle a adoptés.
Article 38
Tarification
des médicaments et critère industriel
Cet article vise à inscrire dans la loi la possibilité de prise en compte de critères d'implantation des lieux de production afin de fixer le prix des médicaments et dispositifs médicaux.
La commission vous demande d'adopter cet article modifié par l'amendement qu'elle a adopté tendant à renforcer la sécurité juridique du dispositif.
I - Le dispositif proposé
A. La mise en oeuvre d'une recommandation du CSIS 2021
Le présent article traduit l'une des mesures du plan « Innovation santé 2030 » présenté en conclusion du conseil stratégique des industries de santé (CSIS) 2021. Il s'agit ainsi de concrétiser l'engagement de « renforcer la prise en compte de l'empreinte industrielle dans la fixation du prix du médicament et des investissements sur notre territoire avec un doublement des crédits CSIS médicaments et leur élargissement aux dispositifs médicaux » et, partant, d'inciter à l'augmentation des capacités de production en vue de l'approvisionnement du marché national .
B. Une possible prise en compte dans les révisions de prix par le comité économique des produits de santé
L'article 38 modifie les articles L. 16216-4 et L. 1652 du code de la sécurité sociale .
Le premier, modifié par le I, précise les modalités de fixation du prix de vente au public pour les médicaments . Le second, modifié par le II, porte des dispositions analogues concernant la fixation du tarif de responsabilité pour les dispositifs médicaux .
Le I comme le II complètent les dispositions des I des deux articles en vue d'y inscrire que la fixation du tarif concerné « peut également tenir compte de la sécurité d'approvisionnement du marché français que garantit l'implantation des sites de production ».
Concernant la conformité du dispositif au droit européen, l'évaluation préalable souligne qu'il conviendra de « justifier, le cas échéant auprès de la Commission européenne, que la mise en oeuvre concrète de ce critère ne constituera pas une distorsion de concurrence au regard du traité sur le fonctionnement de l'Union européenne ou une atteinte à la libre circulation au sein du marché intérieur ».
L'impact financier de la mesure est présenté, au sein de l'évaluation préalable, comme estimé à 15 millions d'euros en 2022 mais des surcoûts annuels de l'ordre de 30 millions d'euros sont attendus .
II - Les modifications adoptées par l'Assemblée nationale
L'Assemblée nationale a adopté cet article sans modification.
III - La position de la commission
A. Le suivi d'une recommandation renouvelée de différents rapports sénatoriaux
La rapporteure soutient le présent dispositif qui répond à une préoccupation impératif de soutenabilité de la production de produits de santé sur le territoire.
Comme a pu le souligner le Leem auprès de la rapporteure, « l'intégration de critères industriels dans la détermination du prix du médicament est importante et pertinente dans le cas de spécialités anciennes dont le prix de vente est aujourd'hui si bas que la question de la viabilité de leur production ou leur approvisionnement sur le sol européen est posée ».
Surtout, la commission souligne qu'il s'inscrit dans la lignée de recommandations sénatoriales renouvelées au cours des récents travaux menés sur le secteur du médicament.
La proposition de loi sénatoriale 411 ( * ) faisant suite en 2019 au rapport de la mission d'information sur la pénurie de médicaments et de vaccins 412 ( * ) , déposée par le sénateur Jean-Pierre Decool, proposait d'inscrire des critères industriels dans la fixation des prix par le CEPS :
- en prévoyant que la révision à la baisse des prix pouvait se faire sur la base de « la soutenabilité des capacités de production de l'entreprise exploitant le médicament et leur adéquation à la demande projetée de la spécialité concernée » ou encore « la place de la spécialité dans l'arsenal thérapeutique disponible sur le territoire français pour le traitement des indications visées » ;
- en donnant à l'agence nationale de sécurité du médicament et des produits de santé la capacité de demander la révision à la hausse du prix de vente pour « tenir compte du risque d'exposition d'un médicament d'intérêt thérapeutique majeur (...) à des ruptures d'approvisionnement récurrentes et des conditions financières et industrielles de son exploitation ».
Plus récemment, dans leur rapport sur l'innovation en santé 413 ( * ) publié en amont des conclusions du CSIS 2021, les sénatrices Annie Delmont-Koropoulis et Véronique Guillotin soulignaient également la nécessité de tenir compte, dans les dépenses consenties en matière de médicament, de la préservation sur le territoire national d'un outil industriel garant d'une meilleure souveraineté sanitaire .
B. Une base légale s'ajoutant à d'autres dispositifs
L'accord-cadre du 5 mars 2021 entre le Comité économique es produits de santé et les entreprises du médicament (Leem) prévoit, à son article 27, une « prise en compte des investissements réalisés dans l'Union européenne ».
Surtout, l'article 28 du même accord concerne directement la question de la sécurité en approvisionnement du marché national. Il prévoit ainsi une possibilité de révision à la hausse du prix au regard des risques sur la production d'une spécialité répondant à un besoin thérapeutique qui ne serait plus couvert.
Une action coordonnée du CEPS en soutien
à la production de médicaments :
l'accord-cadre du
5 mars 2021 entre le CEPS et le Leem
Le nouvel accord-cadre prévoit les dispositions suivantes dans son chapitre III, « soutien aux investissements et aux exportations » :
Article 27 : une stabilité du prix facial de cinq ans maximum peut être accordée par le comité à un produit en fonction des investissements en lien direct avec ce même produit dans les capacités de production, la R&D ou des solutions numériques, récemment réalisés ou à venir en Union européenne, notamment en France (y compris par l'intermédiaire d'un façonnier).
L'industriel s'engage à fournir au comité les montants détaillés, investis ou prévus, les quantités effectivement produites pendant la durée de stabilité ainsi que toute information sur les soutiens publics dont il a bénéficié du fait de ses investissements.
Article 28 : hausse de prix lorsqu'une entreprise fait état d'un risque important pouvant impacter la production ou la commercialisation pour une de ses spécialités pharmaceutiques répondant à un besoin thérapeutique qui ne serait plus couvert au cas où elle disparaitrait du marché . Le comité peut, à l'occasion de la demande de hausse de prix d'une entreprise pour un produit qui aurait des concurrents, se saisir pour un motif de santé publique (notamment pour préserver les capacités d'approvisionnement) d'une révision de prix de tout ou partie d'une classe thérapeutique.
Le bénéfice des dispositions prévues au présent article s'accompagne d'un engagement de l'entreprise à approvisionner le marché français. À défaut, s'il est démontré que la responsabilité de l'entreprise est en cause dans la rupture d'approvisionnement, le comité peut aligner le prix facial hors taxe du médicament sur le prix net.
Article 29 : possibilité de bénéficier des avoirs sur remises au titre du guichet du CSIS pour les entreprises éligibles qui ont réalisé des investissements, dans l'Union européenne et notamment en France, visant en particulier le développement des produits, l'augmentation, l'optimisation ou la digitalisation des capacités de production.
Article 30 : le CEPS peut décider d'une stabilité du prix facial pouvant aller jusqu'à une durée de deux ans renouvelable une fois pour deux ans au plus, pour les produits dont au moins une étape significative de fabrication [principe actif, produit fini, conditionnement] est située en Union Européenne, notamment en France, avec libération des lots effectuée en France et dont plus de 60 % des volumes sont exportés.
Enfin, comme le souligne la direction de la sécurité sociale, en dehors de l'accord-cadre, « les crédits CSIS versés annuellement aux laboratoires ayant investi en France viennent en réduction des remises dues par ces entreprises bénéficiaires, aboutissant, in fine, à une hausse des conditions de prix nets des produits exploités par celles-ci ».
Ainsi, la rapporteure s'interroge sur la portée réelle de la disposition législative proposée, qui n'apparaît pas strictement nécessaire au vu de l'accord-cadre déjà existant, ne semble a priori pas apporter de capacité supplémentaire d'action pour le CEPS ni de contrainte particulière pour la prise en compte de ces critères .
Interrogé sur la redondance éventuelle du présent article avec les stipulations de l'accord cadre et notamment les modalités de prises en compte des investissements, le CEPS considère cependant que article apporte une précision sur le champ de l'avantage en ciblant spécifiquement la sécurisation de l'approvisionnement et la localisation des sites de production.
C. Une formulation qu'il convient de préciser
Si la rédaction du présent article, par son caractère très large, peut sembler très souple, elle n'est pas sans soulever une série de questions.
1. Une mise en oeuvre à préciser
La disposition proposée est bien formulée comme une possibilité de prise en compte et non comme la nécessité de considérer un critère supplémentaire dans la fixation du prix.
En outre, la sécurité d'approvisionnement du marché français n'est pas définie et aucun objectif de stock éventuel ou de durée n'est précisé , pas plus que les critères permettant d'apprécier la garantie que sont censés apporter à ce titre l'implantation des sites de production.
Il apparaît à votre rapporteure que la rédaction retenue cherche à satisfaire un besoin d'affichage du Gouvernement à la suite du CSIS 2021 tout en ménageant la normativité du dispositif pour prévenir les recours qui ne manqueront pas d'être faits au nom du respect des réglementations européennes en matière d'aides d'État.
Ainsi, interrogé sur la mise en oeuvre de cette nouvelle possibilité, le Comité économique des produits de santé a indiqué qu' « il faudra répondre, soit dans un texte soit par la pratique du Comité aux questions de définition du périmètre des produits éligibles (caractérisation du besoin de sécurité d'approvisionnement) en primo inscription, d'identification de la maitrise du risque apportée par l'implantation (que ce soit dans sa localisation ou dans la ou les étapes de la fabrication concernées), de quantification de l'avantage de prix consenti , ou même de suivi de la pérennité de l'implantation et les mesures à prendre en cas de délocalisati on ».
• Ces critères d'objectivation et cet encadrement de l'avantage consenti apparaissent en outre de nature à réduire le risque de contentieux et à assurer le respect du droit européen .
Cependant, comme le constate le CEPS, définir l'avantage de manière objective alors que seul résultat observable est en définitive celui de la négociation globale du prix. Autre élément de complexité alors relevé : quid de l'appréciation de ce critère dans ses conséquences ex post quand le produit valorisé sert de comparateur et de référence à un second ?
2. Différents aspects relevant de l'implantation à mieux appréhender
Par ailleurs, comme le souligne le Leem, concernant les aspects « localisations », certaines situations peuvent soulever des questions de mise en oeuvre de ce critère. Dans le cas par exemple d'une sous-traitance de la production sur le territoire européen, le premier bénéficiaire d'un prix « bonifié » est cependant l'exploitant donneur d'ordre ; il n'est alors pas aisé de s'assurer que le fabricant puisse en bénéficier, au moins partiellement. Le Leem propose à ce titre la possibilité d'un système de convention tripartite entre le CEPS, l'exploitant et le fabricant .
Enfin, l'approche de la localisation doit aussi intégrer la possible complexité de la chaîne de production et des différentes implantations au cours de celle-ci. Sur ce point, le CEPS estime qu'il est possible de distinguer les différentes étapes de la production, « en cohérence avec l'approche de l'accord-cadre dans lequel le Comité a choisi de distinguer les phases de fabrication du principe actif, de fabrication du produit fini et celle du conditionnement » . Celles-ci pourraient conduire à des avantages de niveaux différents.
Le CEPS souligne notamment le cas des dispositifs médicaux. Ainsi, l'un des enjeux sera la capacité du comité à s'assurer que la majeure partie de la valeur du produit est bien produite en France ou en Europe . Si de nombreux « inputs » peuvent être nécessaires à la fabrication d'un dispositif médical (acier, mousse, composants électroniques, etc.) : « il conviendra donc de s'attacher à limiter la dépendance aux fournisseurs internationaux , par exemple en favorisant le "multiple sourcing" ou l'existence de sources d'approvisionnement localisées en Europe, par exemple ».
3. La question particulière des génériques
La rapporteure constate la difficile prise en compte des génériques dans ce dispositif. Si la DSS estime qu'ils sont « éligibles au même titre que les autres produits de santé », le CEPS rappelle que leur tarification est gérée par une disposition spécifique depuis l'accord cadre de 2015 prévoyant une décote uniforme pour tous les acteurs qui vont être concernés par la substitution officinale puis des baisses de prix prévisibles et uniformes. Ainsi, il ne pourra subsister de différence .
Par ailleurs, concernant les dispositifs médicaux , la spécificité d'inscription sous description générique ne permettra pas de valorisation au titre de la localisation .
2. Un encadrement lacunaire et des garanties imprécises
Les valorisations accordées aux termes de l'accord-cadre sont conditionnées, notamment à des conventions précisant les modalités de suivi de la réalisation des engagements en matière d'investissement et de production.
Aucune modalité de contrôle de l'effectivité de ces critères ni aucun engagement de maintien de ces sécurisations de l'approvisionnement ne sont prévus dans le dispositif ici proposé.
La rapporteure estime nécessaire de prévoir un encadrement réglementaire des caractéristiques qui devront être appréciées ainsi que des engagements pris et de leur suivi . Si celui-ci doit ressortir pour partie des pratiques du Comité, il apparaît nécessaire, pour un dispositif pouvant soulever des contentieux importants, de prévoir un encadrement par décret en Conseil d'État .
La commission a ainsi adopté à cette fin l'amendement n° 187.
D. Une préoccupation industrielle qui doit dépasser la seule garantie d'approvisionnement du marché français
Par ailleurs, la rapporteure souligne que le soutien à l'implantation de lignes de production sur le territoire national ou européen ne peut passer par une focalisation sur les seuls médicaments en situation de risque de rupture , comme rappelé dans le rapport 414 ( * ) précité de la commission, sur l'innovation santé.
En effet, comme l'a encore montré la gestion de l'épidémie de covid-19 depuis 2020, la préservation d'une réelle souveraineté sanitaire requiert la présence sur le territoire de chaînes de production nombreuses et potentiellement convertibles . En cela, au-delà même de la question des emplois induits par les industries localisées, il s'agit bien de la présence d'un appareil industriel en capacité d'évoluer.
Aussi, la rapporteure constate que le dispositif du présent article, se bornant à la mention de la sécurisation des approvisionnements, ne doit pas faire oublier tant des médicaments matures sans tension ou risque de rupture, que pour partie les productions de médicaments génériques , et ce alors que les productions de ces deux catégories trouvent une grande pertinence, économique, industrielle et sanitaire, à être maintenues également sur le territoire.
De la même façon, la souveraineté industrielle en matière sanitaire ne peut se concevoir sans une localisation forte d'activités de recherche. Ces aspects demeurent renvoyés à l'accord conclu entre le CEPS et le Leem, qui prévoit de telles valorisations.
Aussi, la rapporteure estime que le critère ici retenu d'implantations de nature à garantir une sécurité d'approvisionnement devra être entendu de manière suffisamment large pour englober ces différents aspects de souveraineté sanitaire.
La commission vous demande d'adopter cet article modifié par l'amendement qu'elle a adopté.
Article 38 bis
(nouveau)
Expérimentation de la prise en charge de substituts
nicotiniques
délivrés par les pharmaciens sans ordonnance
Cet article, inséré par l'Assemblée nationale, vise à autoriser l'expérimentation, pour une durée de deux ans dans trois régions, de la prise en charge par l'assurance maladie de traitements de sevrage tabagique par la dispensation par les pharmaciens d'officine de substituts nicotiniques sans ordonnance.
La commission vous demande d'adopter cet article sans modification.
I - Le dispositif proposé
Résultant de l'adoption par l'Assemblée nationale de deux amendements identiques, l'un du Gouvernement, l'autre de M. Jean-Louis Touraine (La République en Marche), l'article 38 bis du PLFSS pour 2022 vise à permettre à l'État d'autoriser l'expérimentation, pour une durée de deux ans dans trois régions, de la prise en charge par l'assurance maladie des traitements du sevrage tabagique par des substituts nicotiniques qui sont dispensés par les pharmaciens d'officine sans ordonnance ( I ).
Les modalités de mise en oeuvre de l'expérimentation et les territoires concernés seront déterminés par décret. Ce décret devra également préciser les traitements concernés, les honoraires de dispensation du pharmacien définis par la convention nationale 415 ( * ) entre l'assurance maladie et les organisations représentatives des pharmaciens d'officine ainsi que les conditions d'évaluation de l'expérimentation ( II ). Est prévue la possibilité de sélectionner des territoires « contrôles » afin d'évaluer l'impact du dispositif.
Enfin, l'expérimentation devra, à son terme, faire l'objet d'un rapport d'évaluation de l'expérimentation qui devra être transmis au Parlement ( III ).
II - La position de la commission
La commission estime indispensable de renforcer la lutte contre le tabagisme en privilégiant, au-delà des hausses de prix du tabac, une approche proactive en matière de sevrage tabagique. En permettant la prise en charge par l'assurance maladie des substituts nicotiniques délivrés sans ordonnance en officine, l'expérimentation pourrait, à cet égard, contribuer à diminuer la prévalence du tabagisme dans certaines régions où elle reste supérieure à la moyenne nationale.
À titre d'exemple, Santé publique France faisait, en 2019, plusieurs constats préoccupants sur la consommation de tabac dans les Hauts-de-France, dont « une forte prévalence du tabagisme quotidien en Hauts-de-France très liée aux facteurs socio-économiques défavorables de la région », « une plus forte consommation de tabac chez les femmes enceintes » et « un impact sanitaire global de la consommation du tabac particulièrement fort chez les hommes de la région » 416 ( * ) .
Prévalences régionales
standardisées du tabagisme quotidien chez les adultes
de 18 à
75 ans en 2017 (France hexagonale) et en 2014 (outre-mer)
Source : Santé publique France, bulletin de santé publique Hauts-de-France, janvier 2019
Au-delà de la région des Hauts-de-France, la carte de prévalence du tabagisme quotidien publiée par Santé publique France pour 2017 fait apparaître trois autres régions pour lequel cette prévalence est significativement supérieure aux autres régions et au moins supérieure de plus de trois points au taux de prévalence moyen national de 26,9 %. Il s'agit de la Provence-Alpes-Côte d'Azur, de l'Occitanie et du Grand Est. La rapporteure regrette donc que l'expérimentation envisagée par le Gouvernement se limite à trois régions, alors qu'inclure ces quatre régions aurait été plus pertinent en termes de santé publique.
La commission vous demande d'adopter cet article sans modification.
Article 39
Mise en
conformité avec l'obligation de sérialisation
pour les
pharmacies d'officine
Cet article propose de permettre à l'assurance maladie de prononcer des pénalités financières à l'encontre des pharmacies d'officine qui méconnaîtraient leurs obligations en matière de sérialisation des médicaments.
La commission vous demande d'adopter cet article sans modification.
I - Le dispositif proposé
Afin de sécuriser le circuit de distribution des médicaments, la directive 2011/62/UE dite « Médicaments falsifiés » 417 ( * ) et le règlement délégué 2016/161 de la Commission européenne du 2 octobre 2015 418 ( * ) ont institué une obligation de sérialisation des médicaments dispensés par les pharmacies d'officine. Cette obligation se matérialise par la vérification par le pharmacien du numéro d'identification unique de chaque médicament. Encodé dans un code barre appelé « datamatrix », cet identifiant garantit le suivi du médicament tout le long de la chaîne de distribution jusqu'à la dispensation en officine.
Une fois connecté au répertoire national de vérification des médicaments (NMVS) 419 ( * ) , lui-même interfacé avec un répertoire communautaire, le pharmacien doit scanner le code « datamatrix » du médicament afin de vérifier son authenticité. Une fois cette authenticité vérifiée, l'identifiant unique est alors désactivé dans la base afin qu'il ne soit pas attribué à une autre boîte.
Pour mémoire, les pharmaciens ont le choix entre deux solutions pour se connecter au répertoire national : l'une passant par le recours à un connecteur du conseil national de l'ordre des pharmaciens (CNOP), d'un coût de 44 euros hors taxes ; l'autre passant par une connexion manuelle au répertoire par le téléchargement d'un certificat.
Les modalités de connexion des pharmaciens au
répertoire national
de vérification des
médicaments
• Première possibilité : la connexion simplifiée et automatisée à France MVS via le connecteur CNOP :
Cette solution permet de se connecter automatiquement en utilisant l'infrastructure du dossier pharmaceutique (DP), mise en oeuvre par l'ordre. Le connecteur permet d'utiliser les certificats des cartes de professionnels de santé (CPS), déjà utilisés par les logiciels métier des officines, et garantit une connexion en mode pseudonymisé pour l'enregistrement des transactions. Il faut pour cela se munir du courrier reçu de France MVO en février et juin 2020, et se rendre sur : https://fmvo.paynum.fr/ sur lequel sont indiqués les identifiants uniques ; une seule souscription par officine (les pharmaciens titulaires en société' ont le même identifiant) est possible.
Cette solution nécessite de souscrire à France MVO une cotisation annuelle de 44 euros hors taxes par pharmacie ; ce coût est clef en mains et pourrait baisser si toutes les officines sont connectées via le CNOP. Ce système présente un maximum de sécurité et de facilitation pour tous les acteurs et a été acté par les autorités.
La très grande majorité des pharmaciens, par le biais de leurs syndicats, ont souhaité que la connexion au répertoire national de vérification des médicaments passe par le connecteur DP du CNOP afin d'anonymiser les échanges. La mise en place de ce connecteur a nécessité la préparation d'un contrat quadripartite entre l'ordre et France MVO ainsi que leurs prestataires Docaposte et Arvato. Celui-ci a été signé en décembre 2019.
D'autres États membres, en l'occurrence l'Allemagne et l'Espagne, ont également opté pour une connexion des officines via un connecteur développé par leur ordre national des pharmaciens.
• La connexion manuelle :
La connexion manuelle, sans recourir au connecteur du CNOP, requiert une procédure de vérification administrative en plusieurs étapes :
- inscription en tant qu'utilisateur final sur France MVO ;
- communication des pièces justificatives demandées ;
- réception du message électronique envoyé par France MVO comportant les identifiants de connexion ;
- téléchargement du certificat ;
- prise de contact avec l'éditeur de logiciel pour finaliser l'installation du certificat ;
- connexion à France MVS en utilisant les identifiants de connexion et le certificat.
Cette procédure n'est pas automatique et se révèle chronophage, le téléchargement de la photographie de la carte CPS nécessite environ une heure. Il faut renouveler le certificat tous les deux ans. Les pharmaciens doivent se rapprocher de leurs éditeurs pour construire une solution adaptée dont le coût peut varier en fonction de l'opérateur (solution personnalisée, achat d'un certificat d'authentification).
Source : Direction générale de la santé
Selon des informations transmises par la direction
générale de la santé, au 18 octobre 2021, seulement
1 517 officines étaient connectées au répertoire
national de vérification des médicaments, selon France MVO
- organisme national de gouvernance de la sérialisation en
France -, soit 7 % d'officines connectées sur les 21 300
que compte la France. Dans ces conditions, moins de 5 % des
médicaments font l'objet d'une désactivation de leur identifiant
unique.
Concernant les contrôles, les directeurs de santé publique et de l'offre de soins des agences régionales de santé (ARS) ont été sensibilisés sur l'enjeu de la sérialisation des médicaments lors de deux réunions au début du mois de juillet 2021. Ces réunions ont été suivies par la publication le 13 juillet d'une instruction 420 ( * ) qui souligne que « la France est très en retard au niveau de l'Union européenne dans l'application de ce dispositif » et leur demande, en conséquence, d'inclure systématiquement le respect des obligations de la sérialisation par les pharmaciens lors des contrôles en officine et d'adresser avant le 6 septembre un courrier de rappel à la réglementation et aux sanctions encourues à tout titulaire d'officine non connectée au répertoire national de vérification des médicaments.
La direction générale de la santé indique, en outre, que le respect des obligations liées à la sérialisation des médicaments devrait être intégré parmi les objectifs nationaux d'inscription-contrôles (ONIC) à partir de 2022. Un nombre limité d'officines pourrait être inspecté, compte tenu du nombre de pharmaciens inspecteurs de santé publique au sein des ARS et leur implication dans la gestion de la crise covid et dans la vaccination contre la covid-19.
Afin de garantir une mise en oeuvre plus étendue de la sérialisation dans les officines françaises, le I de l'article 39 du PLFSS pour 2022 insère, dans le code de la sécurité sociale, un nouvel article L. 162-16-3-2 prévoyant la possibilité pour le directeur de l'organisme d'assurance maladie territorialement compétent de prononcer une pénalité financière à l'encontre du pharmacien responsable d'officine qui méconnaîtrait ses obligations en matière de sérialisation pour les médicaments remboursables. Le montant de cette pénalité, qui sera déterminé en fonction de la gravité, de la durée et de l'éventuelle réitération des manquements, ne pourra être inférieur à 350 euros, ni excéder, en cumulé, 10 000 euros par année civile.
Ces pénalités seront recouvrées par les unions de recouvrement des cotisations de sécurité sociale et d'allocations familiales (Urssaf) et leur produit affecté à la caisse nationale de l'assurance maladie (CNAM)
Le II de l'article 39 du PLFSS pour 2022 prévoit de limiter, pour le premier mois de mise en oeuvre de cette disposition, l'ampleur des pénalités qui pourraient être prononcées. La pénalité ne pourra ainsi, jusqu'au 31 janvier 2022, être prononcée qu'en cas d'absence totale de connexion au répertoire national de vérification des médicaments et ne pourra excéder 350 euros.
II - Les modifications adoptées par l'Assemblée nationale
Outre un amendement rédactionnel, l'Assemblée nationale a adopté, en première lecture, un amendement du rapporteur général précisant que, pendant la période transitoire jusqu'au 31 janvier 2022, ce sera le montant cumulé de la pénalité qui ne pourra excéder 350 euros.
III - La position de la commission
La commission est attachée à ce que l'ensemble des officines françaises opèrent en conformité avec les obligations européennes applicables en matière de sérialisation et prennent leur part à l'effort collectif de renforcement de la sécurité du circuit de distribution et de dispensation du médicament.
S'il apparaît, en première lecture, surprenant d'inscrire en LFSS des dispositions relatives à la sécurité du circuit de distribution du médicament, sans incidence directe sur l'équilibre financier des comptes sociaux, l'affectation du produit des pénalités aux recettes des organismes d'assurance maladie justifie, selon l'étude d'impact annexée au PLFSS, leur place en loi de financement de la sécurité sociale. Ces recettes supplémentaires, par nature hypothétiques, sont estimées à 1,4 million d'euros en 2022 et à 350 000 euros en 2023, pour s'éteindre à compter de 2024.
La commission vous demande d'adopter cet article sans modification.
* 375 Loi n° 2020-1576 du 14 décembre 2020 de financement de la sécurité sociale pour 2021.
* 376 Article D. 5121-74-1-1 (décret n° 2021-870 du 30 juin 2021 fixant les délais mentionnés aux articles L. 5121-12 et L. 5121-12-1 du code de la santé publique et à l'article L. 162-16-5-4 du code de la sécurité sociale).
* 377 L'obligation des entreprises exploitantes d'assurer la continuité des traitements initiés dans le cadre d'une AAP est inscrite à l'article L. 162-16-5-4 du code de la sécurité sociale.
* 378 L'obligation des entreprises exploitantes d'assurer la continuité des traitements initiés dans le cadre d'une AAC ou d'un CPC est inscrite à l'article L. 162-16-5-2 du code de la sécurité sociale.
* 379 Qui sont précisées par le I bis de l'article L. 162-16-5-4 du code de la sécurité sociale.
* 380 Cette catégorie de médicaments est définie à l'article R. 5121-82 du code de la santé publique.
* 381 Loi n° 2020-1525 du 7 décembre 2020 d'accélération et de simplification de l'action publique.
* 382 Cette liste est visée à l'article L. 162-17 du code de la sécurité sociale.
* 383 En cas d'AMM obtenue après l'inscription sur la liste de rétrocession, le délai de 75 jours court à compter de la délivrance de l'AMM.
* 384 Directive 89/105/CEE du Conseil du 21 décembre 1988 concernant la transparence des mesures régissant la fixation des prix des médicaments à usage humain et leur inclusion dans le champ d'application des systèmes nationaux d'assurance-maladie.
* 385 Rapport de la Commission au Parlement européen et au Conseil COM/2014/0188 final, conformément à l'article 25 du règlement (CE) n° 1394/2007 du Parlement européen et du Conseil concernant les médicaments de thérapie innovante et modifiant la directive 2001/83/CE ainsi que le règlement (CE) n° 726/2004.
* 386 Compte rendu de la séance du 22 janvier 2021 du comité d'interface dédié aux médicaments de thérapie innovante.
* 387 Visée à l'article L. 174-1 du code de la sécurité sociale.
* 388 Loi n° 2008-1330 du 17 décembre 2008 de financement de la sécurité sociale pour 2009.
* 389 Prévue à l'article L. 165-1 du code de la sécurité sociale.
* 390 À savoir, conformément à l'article L. 162-48 du code de la sécurité sociale créé par l'article 24 du PLFSS pour 2022, tout logiciel répondant à la définition du dispositif médical en droit communautaire.
* 391 Mentionnée à l'article L. 165-1 du code de la sécurité sociale.
* 392 « Food and Drug Administration » (FDA).
* 393 Juniper Research, « Digital Therapeutics Market to Exceed $32 billion by 2024 Offering Mixed Fortunes for Drug Companies », communiqué de presse du 15 mai 2019.
* 394 « Portable Document Format ».
* 395 En application de l'article L. 1470-5 du code de la santé publique.
* 396 En application du II de l'article L. 162-16-5-4 du code de la sécurité sociale.
* 397 Résolution européenne n° 69 (2020-2021) sur la conformité au principe de subsidiarité de la proposition de règlement du Parlement européen et du Conseil relatif à un rôle renforcé de l'Agence européenne des médicaments dans la préparation aux crises et la gestion de celles-ci en ce qui concerne les médicaments et les dispositifs médicaux, COM (2020) 725 final.
* 398 Données des rapports annuels d'activité de l'ANSM.
* 399 Dans les conditions prévues au 4° de l'article L. 5126-6 du code de la santé publique.
* 400 C'est-à-dire disposant d'une autorisation de mise sur le marché, d'une autorisation ou d'un cadre de prescription au titre de l'accès compassionnel, d'une autorisation d'importation parallèle ou d'une autorisation d'importation accordée en cas de rupture de stock.
* 401 La recevabilité financière des amendements et des propositions de loi au Sénat , rapport d'information n° 263 (2013-2014) de M. Philippe Marini, fait au nom de la commission des finances, déposé le 7 janvier 2014.
* 402 Délai fixé par la directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain.
* 403 Ce dispositif remplace notamment les dispositifs préexistants d' « autorisation temporaire d'utilisation ». Voir à ce sujet le rapport sur le PLFSS 2021, n° 107, tome II (2020-2021) de M Jean-Marie Vanlerenberghe, rapporteur général, Mme Corinne Imbert, M. René-Paul Savary, Mmes Élisabeth Doineau, Pascale Gruny et M. Philippe Mouiller, déposé le 4 novembre 2020 et le commentaire de l'article 38 relatif à la réforme des dispositifs d'accès dérogatoire aux médicaments.
* 404 La procédure d'inscription aux nomenclatures d'un acte fait l'objet d'une réforme engagée par la LFSS pour 2020 (article 38).
* 405 Réponse du secrétariat d'État auprès de la ministre des solidarités et de la santé publiée dans le JO Sénat du 13/02/2019, page 1091.
* 406 Rapport d'information n° 708 (2020-2021) sur l'innovation en santé.
* 407 État des lieux sur les médicaments biosimilaires - rapport de mai 2016, Agence nationale de sécurité du médicament et des produits de santé.
* 408 Article 42 de la loi n° 2019-1446 du 24 décembre 2019 de financement de la sécurité sociale pour 2020.
* 409 L'interchangeabilité d'un biosimilaire à un autre médicament biologique intervient au moment de la prescription et est à l'initiative du médecin qui échange sur ce point avec son patient. Elle se distingue de la substitution qui a lieu lors de la délivrance à l'initiative du pharmacien.
* 410 Projet de loi de financement de la sécurité sociale pour 2020 - Rapport n° 104 (2019-2020) de M. Jean-Marie Vanlerenberghe, Mme Catherine Deroche, MM. Bernard Bonne, Gérard Dériot, René-Paul Savary et Mme Élisabeth Doineau, fait au nom de la commission des affaires sociales, déposé le 6 novembre 2019, article 29.
* 411 Proposition de loi visant à lutter contre les pénuries de médicaments et de vaccins, n° 463 (2018-2019).
* 412 Rapport d'information de M. Jean-Pierre DECOOL, fait au nom de la MI sur la pénurie de médicaments et de vaccins, n° 737 (2017-2018) - 27 septembre 2018.
* 413 Rapport d'information « Refonder l'écosystème de l'innovation en santé », de Mmes Annie Delmont-Koropoulis et Véronique Guillotin, fait au nom de la commission des affaires sociales, n° 708 (2020-2021) - 23 juin 2021.
* 414 Rapport d'information « Refonder l'écosystème de l'innovation en santé », de Mmes Annie Delmont-Koropoulis et Véronique Guillotin, fait au nom de la commission des affaires sociales, n° 708 (2020-2021) - 23 juin 2021.
* 415 Prévue à l'article L. 162-16-1 du code de la sécurité sociale.
* 416 Santé publique France, bulletin de santé publique Hauts-de-France, janvier 2019.
* 417 Directive 2011/62/UE du Parlement européen et du Conseil du 8 juin 2011 modifiant la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain, en ce qui concerne la prévention de l'introduction dans la chaîne d'approvisionnement légale de médicaments falsifiés
* 418 Règlement délégué (UE) 2016/161 de la Commission du 2 octobre 2015 complétant la directive 2001/83/CE du Parlement européen et du Conseil en fixant les modalités des dispositifs de sécurité figurant sur l'emballage des médicaments à usage humain.
* 419 « National Medicines Verification System ».
* 420 Instruction N° DGS/PP2/2021/151 du 13 juillet 2021 visant à rappeler aux titulaires de pharmacies d'officine leurs obligations prévues par le règlement délégué (UE) 2016/161 de la Commission du 2 octobre 2015 relatif à la sérialisation des médicaments et à en contrôler le respect effectif.